

Key Points
IFN-λ receptor signaling protects against lethal acute GI GVHD.
Recombinant IL-29 treatment in the peri-transplant period promotes gut epithelial regeneration and is a promising therapy to limit GVHD.

Abstract
Immunopathology and intestinal stem cell (ISC) loss in the gastrointestinal (GI) tract is the prima facie manifestation of graft-versus-host disease (GVHD) and is responsible for significant mortality after allogeneic bone marrow transplantation (BMT). Approaches to prevent GVHD to date focus on immune suppression. Here, we identify interferon-λ (IFN-λ; interleukin-28 [IL-28]/IL-29) as a key protector of GI GVHD immunopathology, notably within the ISC compartment. Ifnlr1−/− mice displayed exaggerated GI GVHD and mortality independent of Paneth cells and alterations to the microbiome. Ifnlr1−/− intestinal organoid growth was significantly impaired, and targeted Ifnlr1 deficiency exhibited effects intrinsic to recipient Lgr5+ ISCs and natural killer cells. PEGylated recombinant IL-29 (PEG-rIL-29) treatment of naive mice enhanced Lgr5+ ISC numbers and organoid growth independent of both IL-22 and type I IFN and modulated proliferative and apoptosis gene sets in Lgr5+ ISCs. PEG-rIL-29 treatment improved survival, reduced GVHD severity, and enhanced epithelial proliferation and ISC-derived organoid growth after BMT. The preservation of ISC numbers in response to PEG-rIL-29 after BMT occurred both in the presence and absence of IFN-λ–signaling in recipient natural killer cells. IFN-λ is therefore an attractive and rapidly testable approach to prevent ISC loss and immunopathology during GVHD.
Introduction
Allogeneic bone marrow or stem cell transplantation (hereafter termed BMT) offers cure to patients with high-risk hematologic malignancies. Despite ongoing therapeutic advances, BMT remains limited by transplant-related morbidity and mortality, including that caused by graft-versus-host disease (GVHD). Therapies that prevent GVHD while preserving the graft-versus-leukemia (GVL) effect and pathogen-specific immunity remain a clinical imperative. The gastrointestinal (GI) mucosal barrier is a key target of GVHD and a protective interface between the luminal microbiota and immune cell populations driving lethal acute GVHD (aGVHD).1-4 The manipulation of targets preferentially active at this interface constitutes a rational therapeutic strategy that avoids the detriment of broad immune suppression. Interferons (IFNs) have mixed roles in the GI tract; type II (ie, IFN-γ) are directly pathogenic,5 but type I IFNs can be protective.6,7 Type III IFNs (IFN-λ)8,9 are increasingly recognized as important in controlling immune responses at mucosal surfaces.10-12 We thus sought to define the role of type III IFN-λ in the setting of BMT.
The IFN-λ receptor is a unique heterodimer of the IFN-λ (Ifnlr1) and the interleukin 10 receptor subunit beta (IL-10RB) chains.9 Humans express 4 ligands (IFNL1 [IL-29], IFNL2 [IL-28A], IFNL3 [IL-28B], and IFNL4) and mice express IFNL2 and IFNL3.13-15 IFNλ shares type I IFN downstream signaling pathways, and receptor ligation triggers phosphorylation of STAT1, 2, 3, and 5.9,16 In contrast to the ubiquitous expression of type I IFN receptors, Ifnlr1 is preferentially expressed in mucosal epithelia tissues, especially in the GI and respiratory tracts,11,12,17 and produces distinct transcriptional responses in GI cells.18,19 This compartmentalization of IFN-λ effects in the GI tract suggests mucosal targeting with limited systemic effects.
The role of IFN-λ in innate viral defense was initially shown in hepatitis C infection,20 in which increased rates of spontaneous viral remission and improved responses to therapy were reported in patients with polymorphisms in IFN-λ genes.21 IFN-λ–dependent defense from gastrotropic viruses such as norovirus and rotavirus are also apparent10,11,22 ; however, functions of IFN-λ in nonviral mucosal immunopathology are unexplored. We show that IFN-λ can protect intestinal stem cells (ISCs) and that treatment of BMT recipients with IFN-λ before BMT limits ISC loss and enhances mucosal integrity during GVHD. Together, these data support the use of IFN-λ to prevent GI aGVHD. Because PEGylated recombinant IL-29 (PEG-rIL-29) is in phase 3 clinical trials as an adjunctive therapy for hepatitis C,23,24 a strategy for rapid translation to the clinic can be envisioned.
Methods
Mice
Female mice (≥6 weeks) were derived from strains and sources listed in supplemental Table 1 (available on the Blood Web site). Mice were housed in groups of ≤6 using sterile micro-isolator cages or in co-housing experiments, up to 20 per cage. Housing arrangements were maintained posttransplant, and animals received acidified autoclaved water and standard chow. Water was supplemented with enrofloxacin (100 mg/L) for 2 weeks posttransplantation. No enrofloxacin was supplied in experiments involving fecal microbial 16S sequencing. Tamoxifen (1 mg/d) was administered intraperitoneally for 5 days, 2 weeks before transplant to induce tamoxifen-dependent Cre recombinase activity as indicated. All procedures were approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee or the Fred Hutchinson Cancer Research Center Institutional Animal Care and Use Committee.
Allogeneic stem cell transplantation
Donor bone marrow (BM) was isolated and T cells subsequently removed by using antibody- mediated complement depletion where indicated. T cells were isolated and purified by using magnetic separation. Then, 5 × 106 T cells and 10 × 106 unmanipulated BM cells were transplanted for GVHD-inducing grafts and T cell–depleted BM only for non-GVHD grafts. Cells were introduced intravenously 24 hours after split-dose lethal total body irradiation (TBI); 1000 cGy for B6, 1100 cGy for B6D2F1, and 950 cGy for BM chimeras. GVHD was assessed clinically by using established scoring systems,25 and mice with scores >6 were killed. For GVL experiments, 1 × 106 BCR-ABL nup98hoxA9 or MLL-AF9 transformed leukemia cells were introduced with the graft. Congenic BALB/c or C57Bl/6 strains (CD45.1 and/or CD45.2) were used to identify donor-derived cells in mixed chimeras and in vivo cytotoxicity experiments. For natural killer (NK) cell depletion, anti-NK1.1 antibody (PK136) or mouse immunoglobulin G (IgG) control was given intraperitoneally on day 1 (1 mg per mouse) and days 3 and 6 (0.5 mg per mouse). For PEG-rIL-29 treatment, 5 μg/d was delivered intraperitoneally either 3 days before tissue harvest or on days −2, −1, and 0 of transplantation.
Flow cytometry and antibody staining
Single-cell organ suspensions were prepared by mechanical disruption and lysis where necessary to remove erythrocytes. For isolation and separation of cells from the GI tract before staining and fluorescence-activated cell sorting and/or analysis, the Lamina Propria Dissociation Kit (Miltenyi Biotec) was used as per the manufacturer’s instructions. All analyses were performed on a BD LSR Fortessa flow cytometer (BD Biosciences) unless otherwise specified. Before staining with antibodies detailed in supplemental Table 2, Fc receptor blocking was performed using 2.4G2 (anti-CD16/CD32) incubation for 5 minutes at room temperature. In vivo proliferation assays were performed on T cells labeled with 0.001 mM carboxyfluorescein diacetate succinimidyl ester before transplantation. Intracellular cytokine expression was measured after stimulation with phorbol 12-myristate 13-acetate (PMA) (5 μg/mL) and ionomycin (50 μg/mL) (Sigma-Aldrich) for 4 hours at 37°C, with brefeldin A (BioLegend) included for the last 3 hours of culture. Cells were surfaced labeled, fixed, and permeabilized (Cytofix/Cytoperm Kit; BD Biosciences) and stained with cytokine-specific antibodies. Cell sorting was performed by using a FACSAria II or III (BD Biosciences). Data were processed by using FlowJo version 10 (FlowJo LLC).
Organoid preparation and culture
Organoids were grown from crypt preparations harvested from the small intestine (SI) or colon of naive wild-type (WT) or Ifnlr1−/− mice. Crypt preparations were isolated as previously described26 and growth media derived from the L-WRN (ATCC CRL-3276) cell line (American Type Culture Collection [ATCC]). Fifty viable crypts per well were plated in triplicate, and numbers of growing, viable GI organoids were assessed after 5 days. Four representative fields per well were counted by using the Evos FL inverted microscope, and the average number from 4 fields of each of 3 replicate cultures for an individual crypt donor are reported. Maximal cross-sectional area was calculated by using ImageJ software (National Institutes of Health). Organoids grown from single cell–sorted preparations of Lgr5+ cells were obtained by a modification of the crypt isolation method to generate single-cell suspensions. Fifty Lgr5+ cells were plated per well in triplicate, and total organoids from the entire well were counted to allow assessment of growth efficiency.
Additional methods are included in the supplemental Data. Additional reagents, including chemicals, peptides, and recombinant proteins used in this study, are listed in supplemental Table 3.
Statistical analysis
Survival curves were plotted by using Kaplan-Meier estimates and compared by using log-rank analysis. The nonparametric Mann-Whitney U test (2-sided) was used for comparison between 2 groups, and 1-way analysis of variance with Tukey’s multiple comparison test was used for comparison between 2 or more groups. Data are presented as mean ± standard error of the mean (SEM). Calculations were performed by using Prism 7 for Windows software (GraphPad Software). Additional methods and statistical analysis, including 16S ribosomal sequencing and RNA-sequencing (RNA-seq) gene expression, are outlined in the supplemental Data.
Results
IFN-λ signaling in recipient tissue determines GVHD severity
To examine the role of IFN-λ signaling in BMT, we used B6.Ifnlr1−/− mice (Ifnlr1−/−) that lack the Ifnlr1 receptor subunit and cannot respond to IFN-λ. Fully major histocompatibility complex (MHC)-disparate (BALB/c) BM and T-cell grafts caused early and more severe aGVHD, resulting in enhanced lethality in Ifnlr1−/− recipients (Figure 1A). Clinical GVHD scoring showed more severe disease in Ifnlr1−/− mice (Figure 1B). Weight and GI tract histology of naive Ifnlr1−/− mice did not indicate any developmental abnormalities (supplemental Figure 1); however, semiquantitative histopathology at day 7 post-BMT showed enhanced GVHD in the SI and colon in Ifnlr1−/− mice (Figure 1C-D), suggesting a protective role for IFN-λ. GVHD pathology in the skin and liver were not altered in Ifnlr1−/− recipients (Figure 1D). Ifnlr1−/− recipients also had impaired barrier function in the GI tract (Figure 1E) and displayed systemic dysregulation of the inflammatory cytokines IFN-γ and IL-6 relative to WT recipients (Figure 1F). Total body irradiation resulted in reduced systemic and SI levels of IL-28 (Figure 1G), and although systemic levels of IL-28 remained low after BMT, IL-28 levels increased in the SI during GVHD (Figure 1H). In contrast to the severe GVHD observed in Ifnlr1−/− recipients, Ifnlr1−/− donor grafts did not alter GVHD survival (Figure 1I) or clinical scores (Figure 1J). We confirmed that GVL function of Ifnlr1−/− grafts against MLL-AF9 oncogene-driven primary acute myeloid leukemia (AML) (supplemental Figure 2A) and Philadelphia chromosome–driven blast crisis AML was intact (supplemental Figure 2B). We subsequently focused on the protective mechanisms of IFN-λ signaling in recipient tissues.
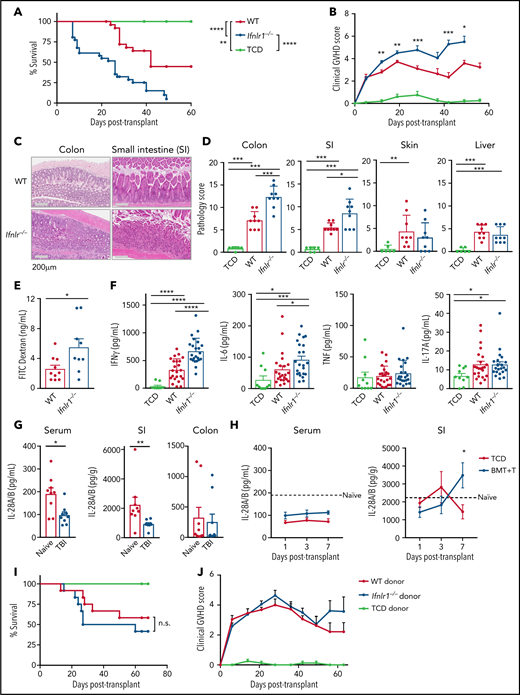
IFN-λ signaling in recipient tissue determines GVHD severity. (A) Survival by Kaplan-Meier estimates for B6.WT (n = 31) and B6.Ifnlr1–/– (Ifnlr1–/–, n = 31) recipient mice lethally irradiated (1000 cGy) and transplanted with BALB/c-derived BM and T cells. A non-GVHD control group received T cell–depleted BM only (TCD; n = 10). Data combined from 5 experiments. (B) Clinical GVHD scores + SEM. (C) Representative images of colon and SI at day 7 after BMT. (D) Semiquantitative GVHD histopathology scores at day 7 after BMT (WT and Ifnlr1–/–, n = 9; TCD, n = 6, combined from 2 experiments). (E) Serum fluorescein isothiocyanate (FITC) dextran at day 7 post-BMT (WT, n = 10; Ifnlr1–/–, n = 9; combined from 2 experiments). (F) Serum IFN-γ, IL-6, tumor necrosis factor (TNF), and IL-17A at day 4 post-BMT (WT & Ifnlr1–/–, n = 23; TCD, n = 10; combined from 3 experiments). (G) IL-28A/B measured in sera, SI, and colon from naive and 24 hours postirradiation (1000 cGy) WT mice (n = 9, combined from 2 experiments). SI and colon mucosal homogenates were prepared, and the IL28-A/B amounts in mucosal supernatants corrected for each gram of tissue. (H) IL-28A/B concentration in serum and SI mucosal homogenates as for panel G at days 1, 3, and 7 after lethal irradiation (1000 cGy) and transplantation with BALB/c BM and T cells or TCD only (n = 9, combined from 2 experiments). (I-J) B6D2F1 recipients were transplanted with BM and T cells from WT or Ifnlr1–/– donors. Survival (I) and GVHD clinical scores (J) (GVHD groups, n = 12; TCD, n = 8; combined from 2 experiments). Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. Kaplan-Meier survival was compared by using the log-rank Mantel-Cox test. *P <.05, **P < .01, ***P <.001, ****P <.0001.
IFN-λ signaling in recipient tissue determines GVHD severity. (A) Survival by Kaplan-Meier estimates for B6.WT (n = 31) and B6.Ifnlr1–/– (Ifnlr1–/–, n = 31) recipient mice lethally irradiated (1000 cGy) and transplanted with BALB/c-derived BM and T cells. A non-GVHD control group received T cell–depleted BM only (TCD; n = 10). Data combined from 5 experiments. (B) Clinical GVHD scores + SEM. (C) Representative images of colon and SI at day 7 after BMT. (D) Semiquantitative GVHD histopathology scores at day 7 after BMT (WT and Ifnlr1–/–, n = 9; TCD, n = 6, combined from 2 experiments). (E) Serum fluorescein isothiocyanate (FITC) dextran at day 7 post-BMT (WT, n = 10; Ifnlr1–/–, n = 9; combined from 2 experiments). (F) Serum IFN-γ, IL-6, tumor necrosis factor (TNF), and IL-17A at day 4 post-BMT (WT & Ifnlr1–/–, n = 23; TCD, n = 10; combined from 3 experiments). (G) IL-28A/B measured in sera, SI, and colon from naive and 24 hours postirradiation (1000 cGy) WT mice (n = 9, combined from 2 experiments). SI and colon mucosal homogenates were prepared, and the IL28-A/B amounts in mucosal supernatants corrected for each gram of tissue. (H) IL-28A/B concentration in serum and SI mucosal homogenates as for panel G at days 1, 3, and 7 after lethal irradiation (1000 cGy) and transplantation with BALB/c BM and T cells or TCD only (n = 9, combined from 2 experiments). (I-J) B6D2F1 recipients were transplanted with BM and T cells from WT or Ifnlr1–/– donors. Survival (I) and GVHD clinical scores (J) (GVHD groups, n = 12; TCD, n = 8; combined from 2 experiments). Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. Kaplan-Meier survival was compared by using the log-rank Mantel-Cox test. *P <.05, **P < .01, ***P <.001, ****P <.0001.
Enhanced GVHD in Ifnlr1−/− recipients is dependent on signaling in hematopoietic and non-hematopoietic cells
To determine if the hematopoietic or nonhematopoietic compartment confers Ifnlr1-driven GVHD protection, we generated BM chimeras lacking Ifnlr1 in each compartment as previously described5,6 (Figure 2A). Chimeric mice were then used as secondary transplant recipients. Ifnlr1 signaling deficiency in either compartment, or both, resulted in enhanced GVHD histopathology in the colon (Figure 2B-C). Serum levels of IFN-γ and IL-6 were elevated in recipients lacking hematopoietic Ifnlr1 signaling only (Figure 2D). These results suggest multiple tissue-specific IFN-λ–driven GVHD protective mechanisms. Ifnlr1 signaling in non-hematopoietic tissues alone seemed to be protective, independent of donor cytokine responses, and thus potentially a consequence of signaling within GI tissues. To confirm this theory, we analyzed Ifnlr1 gene expression in the GI tract and noted high levels of Ifnlr1 messenger RNA in naive GI mucosa (Figure 2E) as previously described.17,27 Indeed, messenger RNA expression of Ifnlr1 in the colon and SI was more than fivefold to 10-fold higher than that in the liver and spleen, suggesting a putative pathway of direct IFN-λ–mediated protection within the epithelium of the GI tract.

Enhanced GVHD in Ifnlr1−/− recipients is dependent on signaling in hematopoietic and nonhematopoietic cells. (A) Transplant schema for creation of BM chimeras and secondary transplants. (B) Representative images of colon at day 7 post-BMT. (C) Semiquantitative colon GVHD histopathology scores at day 7 post-BMT (n = 11 for WT → WT and WT → Ifnlr1–/–, n = 15 for Ifnlr1–/– → WT and n = 14 for Ifnlr1–/– → Ifnlr1–/–, combined from 2 experiments). (D) Serum IFN-γ and IL-6 at day 4 post-BMT. (E) Quantitative polymerase chain reaction enumeration of Ifnlr1 transcripts from naive WT homogenized tissue normalized to expression in liver (n = 3). Data are presented as mean ± SEM. P values were calculated by using analysis of variance and Tukey’s multiple comparison. *P <.05, **P < .01, ***P < .001, ****P < .0001.
Enhanced GVHD in Ifnlr1−/− recipients is dependent on signaling in hematopoietic and nonhematopoietic cells. (A) Transplant schema for creation of BM chimeras and secondary transplants. (B) Representative images of colon at day 7 post-BMT. (C) Semiquantitative colon GVHD histopathology scores at day 7 post-BMT (n = 11 for WT → WT and WT → Ifnlr1–/–, n = 15 for Ifnlr1–/– → WT and n = 14 for Ifnlr1–/– → Ifnlr1–/–, combined from 2 experiments). (D) Serum IFN-γ and IL-6 at day 4 post-BMT. (E) Quantitative polymerase chain reaction enumeration of Ifnlr1 transcripts from naive WT homogenized tissue normalized to expression in liver (n = 3). Data are presented as mean ± SEM. P values were calculated by using analysis of variance and Tukey’s multiple comparison. *P <.05, **P < .01, ***P < .001, ****P < .0001.
Ifnlr1-signaling in recipient NK cells is responsible for the protection from GVHD mediated by hematopoietic cells
We next sought to determine the IFN-λ–responsive hematopoietic cell that attenuates aGVHD and the inflammatory cytokine phenotype. Although no differences in myeloid or lymphoid subset frequencies were observed in naive Ifnlr1−/− mice (data not shown), interrogation of donor T-cell responses after BMT revealed enhanced CD4+ and CD8+ T-cell expansion in Ifnlr1−/− vs WT recipients at day 7 posttransplant (Figure 3A-B). In addition, greater donor T-cell expansion in the spleen (Figure 3C) and colon (Figure 3D) was apparent at day 4 posttransplantation. Donor-derived T cells comprised a greater proportion of the expanding T cells in Ifnlr1−/− recipients (Figure 3E), indicating accelerated donor T-cell expansion. Given that recipient antigen presenting cells (APCs) drive early T-cell expansion posttransplant, we assessed T-cell responses to WT or Ifnlr1−/− dendritic cells (DCs) in mixed lymphocyte cultures. Equivalent CD4+ and CD8+ T-cell expansion was seen when stimulated by WT or Ifnlr1−/− DCs (Figure 3F-G). DC function was also assessed in vivo by using alloantigen-specific CD4+ T-cell responses to donor DCs,28 in which equivalence between WT and Ifnlr1−/− was also observed (Figure 3H-I).
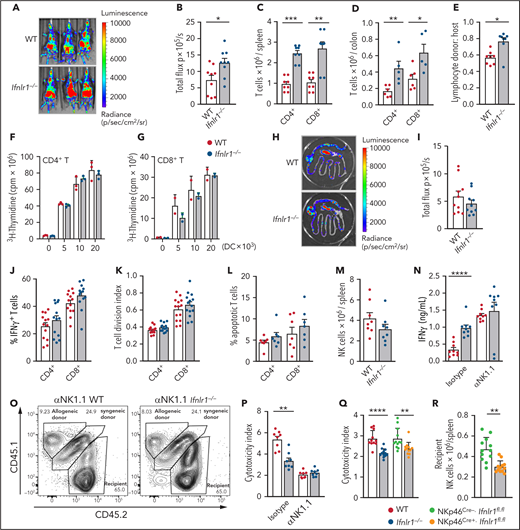
Ifnlr1-signaling in recipient NK cells is responsible for the protection from GVHD mediated by hematopoietic cells. (A-B) Donor BALB/cLuc T-cell expansion in WT and Ifnlr1–/– recipients determined by bioluminescence 7 days post-BMT. Representative images (A) and quantification (n = 9 per group, combined from 3 experiments) (B). (C) Quantification of donor T cells in spleen at day 4 post-BMT (n = 8 per group, combined from 2 experiments). (D) Quantification of donor T cells in colon at day 4 post-BMT (n = 6, combined from 2 experiments). (E) Proportion of T cells at day 4 post-BMT that are donor vs host (n = 8, combined from 2 experiments). (F-G) Functional capacity of splenic DCs from naive B6 WT or Ifnlr1–/– mice to stimulate BALB/c. CD4+ (F) or CD8+ (G) T cells in a mixed lymphocyte reaction (n = 3; data are from 1 of 2 representative experiments). BALB/c recipients were transplanted with WT or Ifnlr1–/– BM + T cells at day 0 and B6.TeaLuc T cells (reactive to BALB/c I-Ed derived TEa peptide expressed in donor B6 I-Ab) were transferred at day 12 to assess donor-derived APC function in the GI tract, as determined by bioluminescence of antigen-specific TEa T cells. (H-I) Representative bioluminescence (H) and quantification (n = 10, combined from 2 experiments) (I). (J) Proportions of donor T cells producing IFN-γ in spleen at day 4 post-BMT (n = 8, combined from 2 experiments). (K) Dividing capacity of splenic BALB/c T cells transplanted into WT or Ifnlr1–/– recipients calculated at day 4 by carboxyfluorescein diacetate succinimidyl ester dilution (n = 14, combined from 3 experiments). (L) Proportion of AnnexinV+7AAD– apoptotic donor T cells at day 4 as in panel K) (n = 7, combined from 2 experiments). (M) Number of NKp46+ cells in naive recipient mice (n = 8). (N) B6 WT or Ifnlr1–/– recipients received αNK1.1 or IgG isotype and were transplanted with BALB/c BM + T cells. IFN-γ was determined in sera at day 4 post-BMT (n = 9 per group, combined from 2 experiments). B6 CD45.2+ WT or Ifnlr1–/– recipients received αNK1.1 or IgG Isotype and were transplanted with CD45.1+ allogeneic BALB/c BM and CD45.1+CD45.2+ syngeneic B6 BM. (O) Representative fluorescence-activated cell sorting plots from NK-depleted recipients showing syngeneic vs allogeneic cells in spleen 48 hours post-BMT. (P) Index of cytotoxicity as described in "Methods" (n = 8, combined from 2 experiments). (Q) Index of cytotoxicity in spleen of WT and Ifnlr1–/– recipient mice in addition to NKp46Cre.Ifnlr1fl.fl-negative and -positive recipients (n = 12, combined from 2 experiments). (R) Number of NKp46+ NK cells in NKp46Cre.Ifnlr1fl.fl-negative and -positive mice 48 hours after 1000 cGy irradiation. Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ifnlr1-signaling in recipient NK cells is responsible for the protection from GVHD mediated by hematopoietic cells. (A-B) Donor BALB/cLuc T-cell expansion in WT and Ifnlr1–/– recipients determined by bioluminescence 7 days post-BMT. Representative images (A) and quantification (n = 9 per group, combined from 3 experiments) (B). (C) Quantification of donor T cells in spleen at day 4 post-BMT (n = 8 per group, combined from 2 experiments). (D) Quantification of donor T cells in colon at day 4 post-BMT (n = 6, combined from 2 experiments). (E) Proportion of T cells at day 4 post-BMT that are donor vs host (n = 8, combined from 2 experiments). (F-G) Functional capacity of splenic DCs from naive B6 WT or Ifnlr1–/– mice to stimulate BALB/c. CD4+ (F) or CD8+ (G) T cells in a mixed lymphocyte reaction (n = 3; data are from 1 of 2 representative experiments). BALB/c recipients were transplanted with WT or Ifnlr1–/– BM + T cells at day 0 and B6.TeaLuc T cells (reactive to BALB/c I-Ed derived TEa peptide expressed in donor B6 I-Ab) were transferred at day 12 to assess donor-derived APC function in the GI tract, as determined by bioluminescence of antigen-specific TEa T cells. (H-I) Representative bioluminescence (H) and quantification (n = 10, combined from 2 experiments) (I). (J) Proportions of donor T cells producing IFN-γ in spleen at day 4 post-BMT (n = 8, combined from 2 experiments). (K) Dividing capacity of splenic BALB/c T cells transplanted into WT or Ifnlr1–/– recipients calculated at day 4 by carboxyfluorescein diacetate succinimidyl ester dilution (n = 14, combined from 3 experiments). (L) Proportion of AnnexinV+7AAD– apoptotic donor T cells at day 4 as in panel K) (n = 7, combined from 2 experiments). (M) Number of NKp46+ cells in naive recipient mice (n = 8). (N) B6 WT or Ifnlr1–/– recipients received αNK1.1 or IgG isotype and were transplanted with BALB/c BM + T cells. IFN-γ was determined in sera at day 4 post-BMT (n = 9 per group, combined from 2 experiments). B6 CD45.2+ WT or Ifnlr1–/– recipients received αNK1.1 or IgG Isotype and were transplanted with CD45.1+ allogeneic BALB/c BM and CD45.1+CD45.2+ syngeneic B6 BM. (O) Representative fluorescence-activated cell sorting plots from NK-depleted recipients showing syngeneic vs allogeneic cells in spleen 48 hours post-BMT. (P) Index of cytotoxicity as described in "Methods" (n = 8, combined from 2 experiments). (Q) Index of cytotoxicity in spleen of WT and Ifnlr1–/– recipient mice in addition to NKp46Cre.Ifnlr1fl.fl-negative and -positive recipients (n = 12, combined from 2 experiments). (R) Number of NKp46+ NK cells in NKp46Cre.Ifnlr1fl.fl-negative and -positive mice 48 hours after 1000 cGy irradiation. Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Assessment of T-cell function showed no increase in the proportion of donor CD4+ or CD8+ T cells expressing IFN-γ (Figure 3J), suggesting that the increased serum IFN-γ seen in Ifnlr1−/− recipients was a consequence of the increased overall T-cell expansion. We next examined donor splenic T-cell proliferation and apoptosis in vivo at day 4 post-BMT and again noted no difference between Ifnlr1−/− and WT recipients (Figure 3K-L). Together, these data suggest that increased donor T-cell expansion in Ifnlr1−/− mice is likely due to enhanced early engraftment (Figure 3E). Because early engraftment is regulated by recipient NK-cell function,29 and IFN-λ–dependent NK function affects tumor models30 and cytomegalovirus infection,31 we interrogated recipient NK cells. Because NK-cell frequencies are unchanged in naive Ifnlr1−/− mice (Figure 3M),30 we used in vivo cytotoxicity assays to assess recipient NK-cell function.29,32 Here, antibody depletion of recipient NK cells with anti-NK1.1 results in accelerated donor T-cell engraftment and enhanced systemic IFN-γ dysregulation in WT recipients at day 4 posttransplant (Figure 3N). This effect was attenuated in Ifnlr1−/− recipients, and recipient NK depletion resulted in equivalent IFN-γ production in WT and Ifnlr1−/− mice. We quantified this functional defect by measuring the relative proportion of MHC-disparate allogeneic or MHC-matched syngeneic targets in the spleen within 48 hours of transplant in the presence or absence of recipient NK cells (Figure 3O-P). When recipient NK cells were depleted, the enhanced anti-donor responses in WT relative to Ifnlr1−/− recipients were lost. To confirm that this effect was intrinsic to Ifnlr1−/− NK cells, assays were repeated by using conditional deletion with NKp46Cre.Ifnlr1fl/fl mice. Here, loss of IFN-λ receptor responses in NK cells recapitulated the impaired anti-donor responses seen in Ifnlr1−/− recipients, with cytotoxic function reduced compared with Cre-negative counterparts (Figure 3Q). We also evaluated numbers of NK cells 48 hours after TBI and found that the absence of IFN-λ–signaling specifically in NK cells reduced their survival (Figure 3R). RNA-seq analysis of NK cells in which the IFN-λ receptor was specifically deleted exhibited differential gene expression–affecting pathways, including apoptosis, consistent with their survival defect after irradiation (supplemental Figure 3; supplemental Table 4). These data identify Ifnlr1-signaling in recipient NK cells as a factor controlling the rate of donor T-cell expansion after BMT.
Ifnlr1-signaling in Lgr5+ ISCs provides nonhematopoietic protection from GVHD
Given the importance of the GI microbiota in determining transplant outcomes,2,33,34 we evaluated the microbiota in WT and Ifnlr1−/− mice by using 16S ribosomal sequencing. Principal component analysis in the 2 strains revealed significant differences at baseline, which coalesced after 4 weeks of cohousing (Figure 4A-B). Cohousing for 4 weeks causes equilibration of microbiota between 2 discrepant in-bred mouse strains.34 Given the differences in the microbiome at baseline and their convergence after cohousing, we undertook BMT in separately and cohoused mice to determine if fecal microbial alterations altered outcome. No differences in posttransplant survival were seen when WT recipients were housed separately or cohoused with Ifnlr1−/− mice or vice versa (Figure 4C). Thus, altered microbiome in Ifnlr1−/− mice was not responsible for enhanced GVHD.

Ifnlr1-signaling in Lgr5+ ISCs provides nonhematopoietic protection from GVHD. (A) Heat map displaying differentially abundant operational taxonomic units consistently increased or decreased in separately housed or cohoused B6 WT and/or Ifnlr1–/– mice. Cohousing was performed for 4 weeks before transplantation (n = 10 per strain and housing condition, combined from 2 experiments). (B) Principal component analysis of fecal microbial composition for mice as in panel A. (C) Survival of recipients in panel A transplanted with BALB/c BM + T cells. (D) Representative images. (E-F) Numbers (E) and size (F) of GI organoids grown from colonic crypt isolates and enumerated at day 5 (n = 6-7, combined from 3 experiments). (G-H) Representative images (G) and semiquantitative GVHD histopathology scores (H) at day 7 after BMT from tamoxifen-treated Cre-positive or Cre-negative Lgr5Cre.Ifnlr1fl.fl recipient mice (n = 10, combined from 2 experiments). (I) Numbers of GI organoids grown from colonic crypt isolates and enumerated at day 5 from mice as in panels D-E (n = 6, combined from 2 experiments). Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. Survival calculated by using the log-rank Mantel-Cox test. *P <.05, **P < .01. PCI, first principal component; PC2, second principal component.
Ifnlr1-signaling in Lgr5+ ISCs provides nonhematopoietic protection from GVHD. (A) Heat map displaying differentially abundant operational taxonomic units consistently increased or decreased in separately housed or cohoused B6 WT and/or Ifnlr1–/– mice. Cohousing was performed for 4 weeks before transplantation (n = 10 per strain and housing condition, combined from 2 experiments). (B) Principal component analysis of fecal microbial composition for mice as in panel A. (C) Survival of recipients in panel A transplanted with BALB/c BM + T cells. (D) Representative images. (E-F) Numbers (E) and size (F) of GI organoids grown from colonic crypt isolates and enumerated at day 5 (n = 6-7, combined from 3 experiments). (G-H) Representative images (G) and semiquantitative GVHD histopathology scores (H) at day 7 after BMT from tamoxifen-treated Cre-positive or Cre-negative Lgr5Cre.Ifnlr1fl.fl recipient mice (n = 10, combined from 2 experiments). (I) Numbers of GI organoids grown from colonic crypt isolates and enumerated at day 5 from mice as in panels D-E (n = 6, combined from 2 experiments). Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. Survival calculated by using the log-rank Mantel-Cox test. *P <.05, **P < .01. PCI, first principal component; PC2, second principal component.
Given that microbiome changes did not contribute to the Ifnlr1−/− aGVHD phenotype, we assessed the role of microbial defenses, specifically Paneth cells and their function. Paneth cells synthesize antimicrobial peptides, are implicated in the maintenance of the GI ISC niche,35 and are reduced in number during aGVHD.36,37 We observed equivalent numbers of Paneth cells post-BMT in WT and Ifnlr1−/− recipients (supplemental Figure 4A-B). We also confirmed equivalent expression of Paneth cell–derived antimicrobial peptides cryptidin, lysozyme T, and regenerating islet-derived protein 3 γ (supplemental Figure 4C). In the absence of IFN-λ–driven protection through microbiota or Paneth cells, we examined the ISC compartment through growth of GI organoids.26,38-40 Organoids are a useful in vitro tool to study GI biology because they recapitulate mature GI mucosal elements and allow manipulation and isolation in real time. We isolated crypts, containing ISCs, from the colon of naive Ifnlr1−/− and WT mice and evaluated growth at 5 days. Ifnlr1−/− crypt donors generated fewer and smaller organoids (Figure 4D-F), implicating Ifnlr1 signaling in maintenance and proliferation of GI epithelia and ISC, from which organoids are grown. To confirm that loss of Ifnlr1 signaling in Lgr5+ISC was relevant to the Ifnlr1−/− GVHD phenotype, we generated Lgr5-EGFP-IREScreERT2.Ifnlr1fl/fl (Lgr5Cre.Ifnlr1fl/fl) mice. Tamoxifen-induced conditional deletion of Ifnlr1 in ISCs in Cre-positive mice resulted in more severe GI aGVHD at day 7 posttransplantation compared with Cre-negative controls (Figure 4G-H). Fewer organoids were also generated from Cre-positive crypt donors (Figure 4I), suggesting that Ifnlr1-signaling responses in the stem cell compartment attenuate GI aGVHD pathology.
IFN-λ treatment produces a proliferative phenotype in GI stem cells
Given that Ifnlr1-signaling seems to mediate protection from GI aGVHD, we tested the therapeutic efficacy of IFN-λ. PEG-rIL-29 has been developed and tested in phase 1 to 3 clinical trials as an adjunctive antiviral therapy for the treatment of hepatitis C virus.23,24,41 It cross-reacts with murine Ifnlr1.30 The capacity of PEG-rIL-29 to support ISCs was assessed by generation of colon organoids from mice receiving PEG-rIL-29 or phosphate-buffered saline (PBS) for 3 days before crypt harvest. Increased numbers of primary colon organoids grew from PEG-rIL-29–treated mice (Figure 5A-B); however, a consistent increase in size was not seen (Figure 5C). Similar, albeit less dramatic, effects were seen in SI organoids (Figure 5D). Organoid growth reflects ISC fitness,42,43 and Lgr5 positivity identifies ISCs.44 We tested PEG-rIL-29 treatment effects on ISCs by using Lgr5-EGFP-IREScreERT2 reporter mice. Although PEG-rIL-29 did not robustly increase numbers of ISC within the SI, there were greater numbers in the colon (Figure 5E). To functionally evaluate PEG-rIL-29–treated ISCs, we flow-sorted single Lgr5+ cells from mice treated with PEG-rIL-29 or PBS for 3 days before ISC harvest and evaluated subsequent organoid growth. Here, we observed that PEG-rIL-29–treated ISCs generated organoids more efficiently than PBS-treated controls (Figure 5F).

IFN-λ treatment produces a proliferative phenotype in GI stem cells. PEG-rIL-29 (5 μg) or PBS was given intraperitoneally for 3 days before gut harvest and evaluation of GI epithelial function. (A-C) Representative images (A), numbers (B), and size of GI organoids (C) grown from colonic crypt isolates (n = 9 per group, combined from 3 experiments). (D-E) Numbers of SI organoids (n = 5, combined from 3 experiments) (D) and number of stem cells (CD45.2–/EpCAM+/GFP+) (E) isolated from digested gut of Lgr5-EGFP-IREScreERT2 mice (n = 5, combined from 3 experiments). (F) Number of organoids grown from FACS sorted single stem cells (n = 4, combined from 3 experiments). (G) Number of colonic organoids from WT and IL-22–/–- mice treated with PBS or PEG-rIL-29 (n = 4, combined from 2 experiments). (H) Number of colon and SI organoids from Ifnar–/– mice treated with PBS or PEG-rIL-29 (n = 6, combined from 3 experiments). (I) RNA-seq from sort purified colonic epithelial cells and stem cells derived from either PEG-rIL-29–treated or PBS-treated mice. Heat map showing top 300 genes significantly differentially expressed in colonic epithelial cells (Lgr5–) and stem cells (Lgr5+) after in vivo PEG-rIL-29 vs PBS treatment (2420 genes total). Expression of the same genes from ISCs derived from PBS- or IL-29–treated mice included for comparison (n = 5 per group). (J) Normalized messenger RNA transcript counts for Ifnlr1, Il10rb, Ifnar1, and Ifnar2 in colonic epithelial cells (Lgr5–) and stem cells (Lgr5+). (K) Functional enrichment analysis of differentially expressed genes: canonical pathway enrichment analysis (log2 fold-change >0.58, and adjusted P value <.05) in sorted Lgr5+ and Lgr5– epithelial cells after in vivo PEG-rIL-29 treatment relative to genotype-matched PBS-treated samples using Ingenuity pathway analysis. Enrichment of canonical pathways associated with immune responses (left) and regulation of cellular proliferation (right). Bubbles represent significant pathway enrichment, as determined by Fisher’s exact test. Bubble diameter represents the log10P value as determined by Fisher’s exact test. Crosses signify a lack of significant pathway enrichment, color indicates predicted pathway activation (red) or predicted inhibition (blue), and white bubbles represent significant functional enrichment of pathways with no available prediction patterns. (L) Venn diagram of overlap of differentially expressed genes in Lgr5+ and Lgr5– cells as for panel K. Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. For RNA-seq, differentially regulated genes were determined after filtering for genes with >5 cpm and fold change differences >1.2 and corrected P values (false discovery rate) <.05. *P <.05, **P < .01, ***P < .001.
IFN-λ treatment produces a proliferative phenotype in GI stem cells. PEG-rIL-29 (5 μg) or PBS was given intraperitoneally for 3 days before gut harvest and evaluation of GI epithelial function. (A-C) Representative images (A), numbers (B), and size of GI organoids (C) grown from colonic crypt isolates (n = 9 per group, combined from 3 experiments). (D-E) Numbers of SI organoids (n = 5, combined from 3 experiments) (D) and number of stem cells (CD45.2–/EpCAM+/GFP+) (E) isolated from digested gut of Lgr5-EGFP-IREScreERT2 mice (n = 5, combined from 3 experiments). (F) Number of organoids grown from FACS sorted single stem cells (n = 4, combined from 3 experiments). (G) Number of colonic organoids from WT and IL-22–/–- mice treated with PBS or PEG-rIL-29 (n = 4, combined from 2 experiments). (H) Number of colon and SI organoids from Ifnar–/– mice treated with PBS or PEG-rIL-29 (n = 6, combined from 3 experiments). (I) RNA-seq from sort purified colonic epithelial cells and stem cells derived from either PEG-rIL-29–treated or PBS-treated mice. Heat map showing top 300 genes significantly differentially expressed in colonic epithelial cells (Lgr5–) and stem cells (Lgr5+) after in vivo PEG-rIL-29 vs PBS treatment (2420 genes total). Expression of the same genes from ISCs derived from PBS- or IL-29–treated mice included for comparison (n = 5 per group). (J) Normalized messenger RNA transcript counts for Ifnlr1, Il10rb, Ifnar1, and Ifnar2 in colonic epithelial cells (Lgr5–) and stem cells (Lgr5+). (K) Functional enrichment analysis of differentially expressed genes: canonical pathway enrichment analysis (log2 fold-change >0.58, and adjusted P value <.05) in sorted Lgr5+ and Lgr5– epithelial cells after in vivo PEG-rIL-29 treatment relative to genotype-matched PBS-treated samples using Ingenuity pathway analysis. Enrichment of canonical pathways associated with immune responses (left) and regulation of cellular proliferation (right). Bubbles represent significant pathway enrichment, as determined by Fisher’s exact test. Bubble diameter represents the log10P value as determined by Fisher’s exact test. Crosses signify a lack of significant pathway enrichment, color indicates predicted pathway activation (red) or predicted inhibition (blue), and white bubbles represent significant functional enrichment of pathways with no available prediction patterns. (L) Venn diagram of overlap of differentially expressed genes in Lgr5+ and Lgr5– cells as for panel K. Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. For RNA-seq, differentially regulated genes were determined after filtering for genes with >5 cpm and fold change differences >1.2 and corrected P values (false discovery rate) <.05. *P <.05, **P < .01, ***P < .001.
Because IL-22 is recognized to support intestinal organoid growth and protection from GI GVHD,45 we tested PEG-rIL-29 treatment in IL-22−/− and WT mice. PEG-rIL-29 enhanced organoid growth equivalently in WT and IL-22−/− strains (Figure 5G), showing that PEG-rIL-29 effects occur independently of IL-22. Similarly, others have shown that GI epithelial barrier integrity is influenced by type I IFNs.7 We report preservation of the PEG-rIL-29 growth advantage in colonic and SI organoids independent of the presence of the type I IFN receptor (Figure 5H).
To explore effects of PEG-rIL-29 in ISCs, we performed RNA-seq transcriptional analysis on purified live CD45.2–, EpCAM+, and Lgr5+and Lgr5–fractions of flow-sorted cells from the digested colon of PBS or PEG-rIL-29 treated mice (supplemental Figure 5). Distinct transcriptional signatures were observed in Lgr5+ and Lgr5– compartments with PBS and PEG-rIL-29 treatment (Figure 5I). The top 25 differentially regulated genes in both the Lgr5+ and Lgr5– compartments are listed in supplemental Tables 5 and 6. Expression of Ifnlr1 and its heterodimeric partner IL10rb was shown in both Lgr5+ and Lgr5− compartments (Figure 5J), complementing expected Ifnar1 and Ifnar2 expression. Canonical pathway enrichment using Ingenuity pathway analysis46 for differentially expressed genes in PEG-rIL-29–treated Lgr5+ and Lgr5– samples, relative to PBS, showed expression of IFN-related genes in both cell types (Figure 5K). Not all genes were shared, however, and some were uniquely upregulated in Lgr5+ cells (Figure 5L). Predictive upstream regulator analyses also identified IFN cytokines (Figure 6A) and IFN regulatory factors (Figure 6B) as drivers of the PEG-rIL-29–induced transcriptional response, and PEG-rIL-29 treatment induced the expression of genes regulating proliferation in Lgr5+ cells. Upstream regulatory analysis identified several recognized regulators of proliferation and cell cycle control (Figure 6C) and transcripts associated with cellular metabolic processes (supplemental Figure 6). These observations complement our functional observations in organoid culture systems (Figure 5A-B), suggesting that although some upregulated pathways are shared in both Lgr5+and Lgr5–cells, effects in organoid-generating Lgr5+ cells contribute to an enhance proliferative capacity. Finally, PEG-rIL-29 induced a distinct type III IFN treatment signature that has previously been identified by Forero et al47 (Figure 6B, highlighted blue), including enrichment of NUPR, SPI, ID2, and ID3 in both PEG-rIL-29–treated Lgr5+ and Lgr5– cells. Together, these data show that PEG-rIL-29 treatment enhances the growth capacity of ISCs, producing type III IFN-specific changes in RNA transcriptional profiles. These effects seem to be independent of IL-22 and type I IFN.

Predicted activation state of PEG-rIL-29–treated Lgr5+ and Lgr5– sorted GI epithelial cells. Cytokines (A), transcriptional regulators (B), and kinases (C) with significantly associated transcriptional changes in sorted Lgr5+ and Lgr5– intestinal epithelial cells after in vivo PEG-rIL-29-administration using Ingenuity pathway analysis. Bubble plot representation of significant enrichment scores (activation z score >2) in at least one treatment condition (ie, PEG-rIL-29). Color indicates predicted activation (red) or predicted inhibition (green), and bubble diameter represents the -log10P value as determined by Fisher’s exact test. Crosses signify a lack of significant activation scores at specific time points.
Predicted activation state of PEG-rIL-29–treated Lgr5+ and Lgr5– sorted GI epithelial cells. Cytokines (A), transcriptional regulators (B), and kinases (C) with significantly associated transcriptional changes in sorted Lgr5+ and Lgr5– intestinal epithelial cells after in vivo PEG-rIL-29-administration using Ingenuity pathway analysis. Bubble plot representation of significant enrichment scores (activation z score >2) in at least one treatment condition (ie, PEG-rIL-29). Color indicates predicted activation (red) or predicted inhibition (green), and bubble diameter represents the -log10P value as determined by Fisher’s exact test. Crosses signify a lack of significant activation scores at specific time points.
IFN-λ treatment protects from GVHD within the GI tract
To test IFN-λ as GVHD prophylaxis in vivo, recipient mice were treated with PEG-rIL-29 from day −2 to day 0 of transplantation. We tested the BALB/c → B6 and B6 → B6D2F1 systems, the latter to illustrate results in the absence of T cell–mediated graft rejection. In both models, PEG-rIL-29 treatment prolonged survival relative to PBS (Figure 7A-B). Systemic inflammatory cytokine dysregulation (Figure 7C) and GI GVHD histopathology were also significantly attenuated in the B6→BALB/c model (Figure 7D-E). No effects were seen in the skin, liver, or lung, and no differences in SI Paneth cell numbers were observed between PBS and PEG-rIL-29 treatment (Figure 7F). We also confirmed that PEG-rIL-29 treatment did not inhibit early hematopoietic engraftment, even in the absence of donor T cells (supplemental Figure 7).
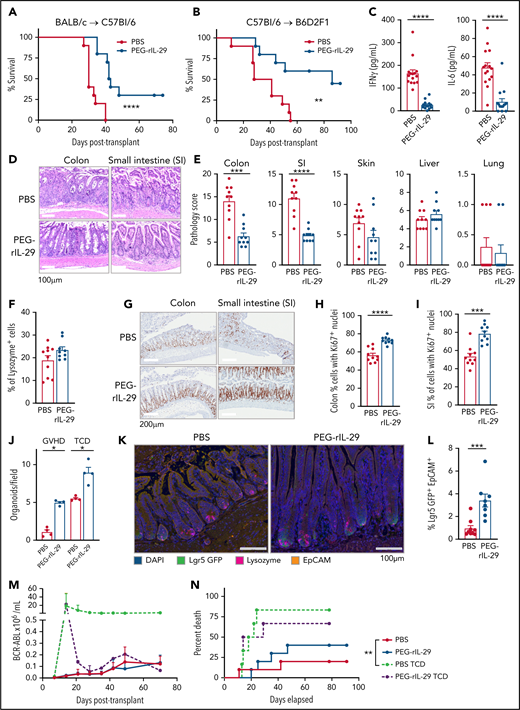
IFN-λ treatment protects from GVHD within the GI tract. PEG-rIL-29 or PBS was given as previously described on days −2, −1, and 0 to BMT recipients. (A) Survival by Kaplan-Meier analysis of B6 recipients transplanted with BALB/c BM + T cells (n = 10, combined from 2 experiments). (B) Survival by Kaplan-Meier analysis of B6D2F1 recipients transplanted with B6 BM + T cells (n = 10, combined from 2 experiments). (C) B6 recipients were transplanted with Balb/c BM + T cells. Serum IFN-γ and IL-6 at day 4 after BMT as in panel A (n = 15, combined from 3 experiments). (D) Representative images of colon and SI at day 7 post-BMT. (E) Semiquantitative GVHD histopathology scores at day 7 post-BMT (n = 10, combined from 2 experiments). (F) Paneth cell numbers. (G) Representative images of proliferation in colon and SI using Ki-67 at day 7 after BMT. (H-I) Quantification of Ki-67 expression at day 7 post-BMT in colon (n = 10, combined from 2 experiments) (H) and in SI (as in panel H) (I). (J) B6 recipients were transplanted with BALB/c BM ± T cells and crypt isolates obtained 7 days later. Colon organoids were quantified (n = 4, combined from 2 replicate experiments). (K-L) Representative images (K) and enumeration (L) of Lgr5+ ISCs in tissue sections at day 7 post-BMT from ileum from PBS or PEG-rIL-29 recipients of BALB/c BM + T cells after NK-cell depletion with NK1.1 on day −1 (1 mg), day +3 (0.5 mg), and day +6 (0.5 mg), (n = 9, combined from 2 experiments). (M) B6D2F1 recipients were treated with PBS or PEG-rIL-29, then transplanted with BM ± T cells from B6.WT donors, together with recipient type BCR-ABL nup98hoxA9 leukemia expressing GFP. (M) The number of GFP+ leukemia cells was determined in peripheral blood thereafter (n = 10, combined from 2 experiments). (N) Death from leukemia in recipients transplanted as in panel M. Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. Survival was calculated by using the log-rank Mantel-Cox test. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; TCD, T cell–depleted.
IFN-λ treatment protects from GVHD within the GI tract. PEG-rIL-29 or PBS was given as previously described on days −2, −1, and 0 to BMT recipients. (A) Survival by Kaplan-Meier analysis of B6 recipients transplanted with BALB/c BM + T cells (n = 10, combined from 2 experiments). (B) Survival by Kaplan-Meier analysis of B6D2F1 recipients transplanted with B6 BM + T cells (n = 10, combined from 2 experiments). (C) B6 recipients were transplanted with Balb/c BM + T cells. Serum IFN-γ and IL-6 at day 4 after BMT as in panel A (n = 15, combined from 3 experiments). (D) Representative images of colon and SI at day 7 post-BMT. (E) Semiquantitative GVHD histopathology scores at day 7 post-BMT (n = 10, combined from 2 experiments). (F) Paneth cell numbers. (G) Representative images of proliferation in colon and SI using Ki-67 at day 7 after BMT. (H-I) Quantification of Ki-67 expression at day 7 post-BMT in colon (n = 10, combined from 2 experiments) (H) and in SI (as in panel H) (I). (J) B6 recipients were transplanted with BALB/c BM ± T cells and crypt isolates obtained 7 days later. Colon organoids were quantified (n = 4, combined from 2 replicate experiments). (K-L) Representative images (K) and enumeration (L) of Lgr5+ ISCs in tissue sections at day 7 post-BMT from ileum from PBS or PEG-rIL-29 recipients of BALB/c BM + T cells after NK-cell depletion with NK1.1 on day −1 (1 mg), day +3 (0.5 mg), and day +6 (0.5 mg), (n = 9, combined from 2 experiments). (M) B6D2F1 recipients were treated with PBS or PEG-rIL-29, then transplanted with BM ± T cells from B6.WT donors, together with recipient type BCR-ABL nup98hoxA9 leukemia expressing GFP. (M) The number of GFP+ leukemia cells was determined in peripheral blood thereafter (n = 10, combined from 2 experiments). (N) Death from leukemia in recipients transplanted as in panel M. Data are presented as mean ± SEM. P values were calculated by using the 2-tailed Mann-Whitney t test. Survival was calculated by using the log-rank Mantel-Cox test. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; TCD, T cell–depleted.
We next examined in vivo proliferative effects of PEG-rIL-29 treatment on ISCs post-BMT. Ki67 expression was significantly higher in colon and SI epithelial cells, including crypt bases where stem cell reside, showing that pretransplant PEG-rIL-29 drives epithelial proliferation post-BMT (Figure 7G-I). To examine ISC fitness, we quantified colonic organoid growth from crypt isolates 7 days after BMT from mice with or without GVHD. As expected, GVHD impaired intestinal organoid generation, whereas PEG-rIL-29 treatment of recipients improved organoid formation with and without GVHD (Figure 7J). We then enumerated ISC posttransplant after PBS or PEG-rIL-29 treatment in a BALB/c to B6 model in which recipient NK cells were depleted pretransplant; the goal was to examine the dependence of the PEG-rIL-29 treatment effect on ISC numbers in vivo in the absence of any NK-dependent effect. Increased numbers of ISCs were observed by multispectral imaging on day 7 after PEG-rIL-29 treatment (Figure 7K-L), with no alteration in Paneth cell numbers, proximity of ISCs to Paneth cells, or numbers of infiltrating T cells in the GI tract (supplemental Figure 8A-D). Quantitation of ISC numbers by using flow cytometry after GI tissue digestion showed similar trends after PEG-rIL-29 treatment in NK-replete recipients, confirming this effect was independent of effects of IFN-λ in recipient NK cells (supplemental Figure 8E-F). Dual immuno-fluorescent labeling revealed co-staining of Ki67 and Lgr5+, consistent with the increases in Ki67 previously shown broadly in GI mucosa (supplemental Figure 8G). Together, these data functionally validate the PEG-rIL-29–mediated upregulation of proliferative pathways in Lgr5+ cells seen in the RNA-seq analysis. Finally, we confirmed that PEG-rIL-29 treatment did not impair GVL using GFP-expressing BCR-ABL nup98HoxA9 AML. Treatment did not influence leukemia growth or leukemic death in mice relative to PBS controls (Figure 7M-N).
Discussion
We have shown dual roles for IFN-λ in the prevention of GI aGVHD through both hematopoietic and non-hematopoietic recipient cells. We observed impaired NK-cell function in the absence of Ifnlr1 signaling, resulting in accelerated donor T-cell engraftment. We also noted IFN-λ–dependent improvements in intestinal epithelial barrier and ISC function, at least in part via effects on gut epithelium. Critically, we showed that these effects can be exploited therapeutically, as PEG-rIL-29 administration limited ISC loss and protected from severe GI aGVHD.
IFN-λ is a key effector cytokine of mucosal immunity, with others exhibiting a unique and nonredundant role in immune defense at mucosal barriers,10,11,27,48-50 which is supported by our findings in the context of GVHD. In the absence of effective immunologic interventions to separate GVHD and GVL, protecting key tissue targets from immunopathology is an attractive alternative.51 Transplantation and GVHD cause disruption to the integrity of this barrier, preservation of which is critical to the prevention of GVHD lethality.28,52 IFN-λ treatment directly improves the proliferative and regenerative capacity of mature GI epithelia through Lgr5+ ISCs, thus promoting epithelial barrier integrity. Although we showed preservation of Lgr5+ ISC numbers after PEG-rIL-29 treatment, our transcriptomic analysis suggests responses occur in both ISC and mature epithelial cell compartments, and both may contribute to the protection from GVHD. Our findings here are in contrast with a recent study that did not identify reductions in GVHD mortality in Ifnlr1−/− recipients after BMT in a single transplant system.53 Unfortunately, the absence of specific data from this prior study, including clinical and particularly histologic assessment of GVHD in the GI tract, makes interpretation difficult. Although the investigators did administer a locally generated PASylated cytokine, the nature of the cytokine (IL-29 vs IL-28), the administration schedule, and activity of the PEGylated clinical grade recombinant IL-29 used in our studies were significantly different. Importantly, PEG-rIL-29 administration, as well as both broad and lineage-specific Ifnlr1 deletion, produced consistent and reproducible effects on ISC survival in our studies.
We confirm and extend the findings of Souza-Fonseca-Guimaraes et al30 showing NK-cell modulation by IFN-λ, now indicating that these are direct rather than indirect effects and that they confer a pro-survival function after radiation. Others have described additional effects in DCs and neutrophil subsets,30,54-57 together with tissue-specific innate antiviral effects10-12,22,49,58 that are consistent with the described efficacy of IFN-λ in the treatment of hepatitis C.20,59-62 Together, these findings highlight roles for IFN-λ in both hematopoietic and non-hematopoietic compartments. In our model, IFN-λ signaling in recipient NK cells seemed to modestly slow the initial rate of T-cell engraftment and consequent systemic inflammatory cytokine generation that indirectly limit GVHD target tissue damage. These effects likely compound the direct influence mediated by the preferential expression of IFN-λ receptors on tissues of epithelial origin.63 Our findings in GVHD show the IFN-λ–mediated indirect hematopoietic and direct non-hematopoietic effects converging on GI epithelia and ISCs.
Manipulating cytokine signaling is a rational strategy to alter immune-mediated pathologies, including GVHD.45,64-66 To date, cytoprotection within the GI tract has been explored in relation to IL-11 and keratinocyte growth factor.67-70 The latter showed clinical activity in limiting the severity of mucositis in chemoradiotherapy-conditioned transplant patients, but this did not translate to the attenuation of aGVHD.71,72 More recently, IL-22 has been shown to preserve Paneth cell numbers and antimicrobial peptide secretion and to protect the stem cell niche, resulting in attenuation of aGVHD in mice.45,73-75 However, IL-22 has also been shown to mediate cutaneous GVHD,76 and therefore treatment with this cytokine may have undesired effects. Importantly, we show that the effects mediated by IFN-λ are independent of IL-22. Recipient-derived IL-17A has also been shown to be important in preventing dysbiosis and maintaining intestinal barrier function after BMT, an effect partially mediated by mucosal-associated invariant T cells.34,77 Thus, cytokines in the IFN and IL-17/IL-22 family seem important and clinically useful to enhance intestinal barrier function.50,78 Importantly, however, IFN-λ does not seem to cause pathology within the hematopoietic compartment; specifically, there seem to be no effects on antigen-presenting cells or alterations to T-cell function that impair GVL or pathogen-specific immunity.
The availability of clinical grade IFN-λ with an established safety profile makes our findings rapidly translatable and testable in the clinic. There is also clear potential for the use of IFN-λ outside of BMT, particularly in other immunopathologies that cause specific damage to GI epithelia such as inflammatory bowel disease.63,79 Regeneration of epithelial components in ulcerative colitis would rationally limit morbidity, and links to disease severity and luminal microbial content have been made in Crohn’s disease.80,81 Reduction of morbidity associated with therapeutic radiation of fields encompassing the GI tract is an additional logical application for the effects seen in our experiments. Similarly, specific GI tract protection from damage as a consequence of intensive chemotherapy regimens, such as those during induction therapy for AML, is also testable. Our data provide a sound basis for the therapeutic exploitation of IFN-λ–mediated protection of the GI epithelia in allogeneic BMT that avoids off-target effects on leukemia and pathogen-specific immunity.
Acknowledgments
The authors thank Leanne Cooper for her assistance in preparation of samples for RNAseq and the QIMR Berghofer Medical Research Institute Core Flow Cytometry and Histology departments. A.S.H. was supported by a Leukemia Foundation clinical scholarship, The Royal Brisbane and Women's Hospital (RBWH) Foundation, and a Haematology Society of Australia and New Zealand New Investigator Scholarship. The experimental work was supported by grants from the National Health and Medical Research Council (APP1146859). B.R.B. was supported by that National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (R01 HL56067 and R01 HL155114) and the NIH/National Institute of Allergy and Infectious Diseases (R37 AI34495). G.R.H. was supported by the NIH/National Heart, Lung, and Blood Institute (R01 HL148164). Experimental histopathology at Fred Hutchinson Cancer Research Center was supported by the NIH/National Cancer Institute (P30 CA015704). Scientific Computing Infrastructure at Fred Hutchinson Cancer Research Center was supported by Office of Research Infrastructure Programs (ORIP) grant S10OD028685.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: G.R.H., A.S.H., and K.H.G. were responsible for conceptualization; A.S.H., G.R.H., P.H., and J.B. were responsible for methodology; K.L.B., A.S.H., S.H.K., K.H.G., and M.K. were responsible for software; A.S.H., M.K., R.J.R., A.F., R.D.K., K.S.E., K.C., A.V., R.G., and K.L.B. were responsible for investigation; P.H., N.W., R.S., S.W.L., M.A.D.-E., S.V.K., and J.B. provided resources; A.S.H., M.K., A.D.C., K.H.G., S.H.K., and K.L.B. conducted the formal analysis; A.S.H. and K.H.G. were responsible for data curation; A.S.H. wrote the original draft; B.R.B., G.R.H., and K.H.G. wrote, reviewed, and edited the manuscript; and G.R.H., K.H.G., B.R.B., and S.W.L. were responsible for study supervision.
Conflict-of-interest disclosure: A.S.H., M.K., K.H.G., and G.R.H. are inventors on patents describing methods to attenuate immune pathology in the GI tract that includes IFN-λ. S.V.K. is an inventor on patents and patent applications related to IFN-λs, which have been licensed for commercial development. G.R.H. has consulted for Generon Corporation and NapaJen Pharma; and receives research funding from Compass Therapeutics, Syndax Pharmaceuticals, Applied Molecular Transport, and iTeos Therapeutics. B.R.B. receives remuneration as an advisor to Magenta Therapeutics and BlueRock Therapeutics; research funding from BlueRock Therapeutics and Rheos Medicines; and is a co-founder of Tmunity Therapeutics. K.H.G. has consulted for, and receives research funding, from CSL Ltd. The remaining authors declare no competing financial interests.
Correspondence: Geoffrey R. Hill, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Seattle, WA 98109; e-mail: grhill@fredhutch.org.
The 16S microbial sequencing data have been submitted to the NCBI Sequence Read Archive under BioProject PRJNA507914.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal