

In this issue of Blood, characterize 2 microRNAs (miRNAs) that are effectors of malignant transformation by the MLL-AF4 fusion protein.1 Mixed lineage leukemia (MLL) gene fusions are the molecular hallmark of infant acute lymphoblastic leukemia (ALL) and are present in tumor cells in up to 80% of patients.2 Detection of the gene fusion in neonatal blood spots indicates that MLL-rearranged infant ALL originates in utero. The small number of potentially cooperating genetic lesions suggests that MLL-AF4 is sufficient to induce infant ALL.3 Detection of the MLL-AF4 fusion protein in CD34+/CD19− fetal hematopoietic stem or progenitor cells or in fetal cells before hematologic specification suggests that there is a window of opportunity during development for the MLL-AF4 fusion protein to immortalize hematopoietic stem and progenitor cells (HSPCs). In an earlier study, Barrett et al used a conditional invertor mouse line for cell-specific expression of an Mll-AF4 fusion protein. Targeting of Mll-AF4 in early fetal HSPCs enhanced the lymphoid potential of primed multipotent cells but was insufficient by itself to induce leukemia.4
Malouf et al found 2 miRNAs (miR-128a, miR-130b) that were significantly upregulated in tumor cells from pediatric MLL-AF4+ ALL patients.1 MLL-AF4 binding upstream of the miR-128a and miR-130b transcriptional start sites suggested direct regulation. Knockdown of miR-128a and/or miR-130b significantly impaired proliferation of human MLL-AF4+ cell lines, and inhibition of miR-130a significantly impaired disease initiation by the pediatric MLL-AF4+ SEM ALL cell line in immunodeficient NOD-scid IL2Rgammanull (NSG) mice. To address the role of these miRNAs in transformation, the investigators overexpressed them in lineage-marker–depleted Sca1+Kit+ (LSK) fetal liver cells from targeted Mll-AF4 invertor mice (see figure). Both miRNAs increased the clonogenic B-cell potential of Mll-AF4+ cells. Transplantation of Mll-AF4+ LSK cells virally expressing miR-128a (pMIRH-128a) or miR-130b (pMIRH-130b) induced a hematologic malignancy in mice with increasing penetrance upon propagation into secondary and tertiary recipients. pMIRH-130b mice presented with a mixed B-cell/myeloid malignancy, whereas pMIRH-128a mice developed pro–B-cell ALL. Similar to MLL-AF4+ human ALL, pMIRH-128a– and pMIRH-130b–transplanted mice showed significant leptomeningeal tumor cell infiltration. Comparative transcriptomic analysis of tumor cells from diseased mice revealed the expression of several well-known MLL-rearranged leukemia-associated target genes, including Meis1, Runx1 (pMIRH-128a mice), HoxA9, Flt3 (pMIRH-130b mice), Cdk6, and Bcl2. Knockdown experiments showed that, although miR-128a seemed to be critical for initiation of the disease, miR-130b was essential for its maintenance. Further transplantation experiments demonstrated that lympho-myeloid progenitors (LMPPs) allowed the propagation of pMIRH-130b mixed B-cell/myeloid leukemia, whereas Lin-Sca1+/−Il7+Kit+ blasts maintained pro–B-ALL disease of pMIRH-128a mice. Malouf et al also identified several candidate target genes that may mediate the leukemic activity of miR-128 (including the nuclear orphan receptor NR2F6 gene), miR-130b, or both (including the sphingomyelin synthase 1 [SGMS1] gene). NR2F6 and SGMS1 expression was downregulated in tumor cells of pMIRH-128a and pMIRH-130b mice; overexpression of these genes decreased proliferation or induced apoptosis of human MLL-AF4+ ALL cell lines, respectively. Transplant experiments in which miRNA knockdown was combined with target overexpression suggest that induction, as well as maintenance, of the disease phenotype is miRNA dependent and involves downregulation of NR2F6 and SGMS1. Collectively, Malouf et al identified 2 MLL-AF4–regulated and functionally relevant miRNAs (miR-128a, miR-130b) targeting 2 critical downstream effectors that contribute to malignant transformation of fetal liver–derived hematopoietic cells.
Previous studies identified several aberrantly expressed miRNAs in MLL-AF4+ ALL. Notably, some miRNAs seem to control expression of the MLL-AF4 fusion protein, but no miRNA has been functionally validated as a critical fusion protein downstream target.5 In addition, being a potential ALL biomarker, miR128a was characterized as a regulator of stemness and differentiation of leukemic cells.6 This biological activity seems to be supported, in part, by the findings of Malouf et al: miR-128a maintained stemness and completely blocked the differentiation of Mll-AF4+ fetal HSPCs at the pro–B-cell state. miR-130b has previously been linked to malignant transformation by the human T-cell leukemia virus 1 by protecting it from apoptosis.7 Similarly, Malouf et al found that blocking miR-130b impaired proliferation and induced apoptosis of human MLL-AF4+ ALL cell lines.
Among multiple predicted miR-128a and miR-130b targets that are differentially expressed in human MLL-AF4+ ALL, Malouf et al functionally validated downregulation of NR2F6 and SGMS1 as mediators of induction, as well as maintenance of, Mll-AF4–driven disease. Previous studies showed that NR2F6 overexpression in murine bone marrow cells increased colony formation, induced myelodysplasia, and favored leukemic myeloid transformation, suggesting lineage-specific activity.8 SGMS1 catalyzes the synthesis of sphingomyelin, a sphingolipid that is an important component of membrane microdomains (aka lipid rafts) and modulates transmembrane signals. SGMS1 was previously found to be a proliferation mediator of the BCR-ABL fusion protein and was upregulated in myeloid leukemia upon exposure to chemotherapeutic agents.9 Therefore, the observations by Malouf et al suggest that transformation of fetal hematopoietic cells by MLL-AF4 depends on the particular, but not yet determined, sphingolipid composition of the cellular membrane microdomains. It will be interesting to see how small molecules that selectively increase or inhibit SGMS1 expression and/or activity may modulate the cellular-transforming activity of the MLL-AF4 fusion protein.
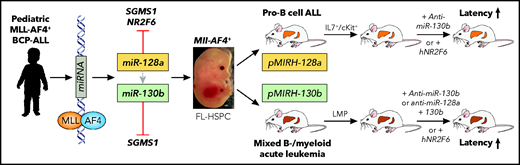
Functional in vivo characterization of MLL-AF4–regulated miRNAs. miR-128a and miR-130b were identified as MLL-AF4 targets in tumor cells from patients with pediatric B-cell precursor (BCP) ALL. Reconstitution of mice with fetal liver(FL)-derived Mll-AF4+ HSPCs induced a pro–B-cell ALL (miR-128a) or a mixed B-cell/myeloid acute leukemia (miR-130b) propagated by Il7r+/c-Kit+ or LMPP leukemic blasts, respectively. Specific miRNA inhibition or overexpression of hNR2F6 impaired leukemia propagation. Professional illustration by Patrick Lane, ScEYEnce Studios.
Functional in vivo characterization of MLL-AF4–regulated miRNAs. miR-128a and miR-130b were identified as MLL-AF4 targets in tumor cells from patients with pediatric B-cell precursor (BCP) ALL. Reconstitution of mice with fetal liver(FL)-derived Mll-AF4+ HSPCs induced a pro–B-cell ALL (miR-128a) or a mixed B-cell/myeloid acute leukemia (miR-130b) propagated by Il7r+/c-Kit+ or LMPP leukemic blasts, respectively. Specific miRNA inhibition or overexpression of hNR2F6 impaired leukemia propagation. Professional illustration by Patrick Lane, ScEYEnce Studios.
Although this impressive piece of work provides novel pieces of the puzzle with regard to MLL-AF4–driven leukemogenesis, the study by Malouf et al also illustrates its complexity. There are most likely additional miRNAs beyond miR-128a and miR-130b, as well as other downstream effector targets that all somehow contribute to the transforming activity of the MLL-AF4 fusion protein. Future studies are necessary to dissect the critical signaling nodes for targeted interference; however, most importantly, we need to identify strategies to efficiently inactivate the MLL-AF4 fusion protein itself, which appears to be the conductor of an orchestra that drives the biology of this deadly disease.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal