

In this issue of Blood, 1 identify 3 unrelated individuals with Shwachman-Diamond syndrome (SDS) with compound, pathogenic, heterozygous, elongation-factor–like GTPase-1 (EFL1) variants.
First, their findings reward their doggedness in seeking a clinically suspected genetic etiology. They observed 3 suspected SDS cases. Initially described in the 1960s, SDS typically presents with chronic neutropenia, exocrine pancreatic insufficiency, bone lesion(s), and/or developmental delay, all associated with an incredibly high rate, ∼30%, of secondary leukemic transformation.2 The identification, ∼ 20 years ago, of deleterious biallelic SBDS variants as the cause of most cases of SDSs3 stimulated relentless investigation into the function of SBDS and its role in disease. This work demonstrated that SBDS and GTPase EFL1 participated together in evicting antiassociation factor eIF6 from the nascent large 60S ribosomal subunit, enabling its fusion with 40S, which yielded the mature 80S ribosomes responsible for translation. Hence, SDS was classified as a ribosomopathy, with defective mature ribosome production.4 Recent studies5,6 have shown that deleterious, biallelic germline EFL1 variants represented a previously undescribed SDS etiology and demonstrate the combined role of SBDS and EFL1 in the last step of ribosome maturation.5,6
Here, the authors first identified a single germline, pathogenic EFL1 variant, in 3 SDS patients (https://www.biorxiv.org/content/10.1101/483362v1.full.pdf), but as EFL1 deficiency was previously reported to have autosomal-recessive inheritance,5,6 finding the second pathogenic event was crucial to understanding the clinical features observed. After further painstakingly meticulous genetic investigations, the authors identified the second germline pathogenic EFL1 variant, showing all 3 patient with SDS carried compound, heterozygous, germline, pathogenic EFL1 variants.
The difficulty the authors encountered in detecting one of the EFL1 variants was due to the existence of several genomic sequences closely homologous to the EFL1 gene, including 2 EFL1 pseudogenes (EFL1P1 and EFL1P2) and segmental duplications.6SBDS, which also possesses a pseudogene (SBDSP1) with high sequence homology, has a strikingly similar detection difficulty.3
The features mentioned above make genetic diagnosis of SDS challenging. In the present study, another genetic event complicated the analysis and interpretation of the molecular etiology of SDS. Indeed, Lee et al observed disequilibrium of EFL1-variant frequencies in the 3 patients’ blood cells. Although each variant’s expected variant-allele frequency (VAF) in the context of compound heterozygosity is ∼ 50%, the EFL1 variant’s VAF was markedly underrepresented in the patients’ blood cells (8.3%, 14.8%, and 36.8%), whereas the VAF of the pathogenic alleles in the patients' parents with available data was ∼50%, as expected in the context of heterozygosity.
The authors demonstrated that the disequilibrium of EFL1-variant allele frequencies in patients' hematopoietic cells resulted from a somatic uniparental disomy (UPD) leading to neutral copy number–loss of heterozygosity (CN-LOH) of all or part of chromosome 15 harboring the EFL1 locus. Functional analysis of EFL1-knockout cell lines reconstituted with the mutated EFL1 forms, and studies in animal models (zebrafish and mice) indicated that one of the EFL1 variants identified in the patients was functionally more deleterious than the other.
Remarkably, the somatic UPD led to replacement of the EFL1 allele carrying the more damaging variant by the less deleterious one, rendering the cells with the somatic UPD homozygous for the less detrimental EFL1 variant. That trait gives them a selective advantage over nonmodified cells and promotes their clonal expansion. When sought in several organs, UPD detection was limited to the blood compartment, suggesting that the somatic CN-LOH occurred only in the patients’ hematopoietic system (see figure).
Second, beyond demonstrating the role of segmental UPD of EFL1 in SDS, this work opens new perspectives. Somatic genetic rescue (SGR; ie, somatic genetic modifications that fully or partially counteract the deleterious effects of germline mutations, thereby providing a selective advantage to the unmodified cells) has been detected in other Mendelian hematopoietic diseases. In these rare cases, SGR has been shown to substantially blunt the patients’ clinical features and even completely cure the disease.7
In this study, the UPD CN-LOH can be considered a partial SGR, resulting in blood compartment mosaicism. Although the frequencies of UPD CN-LOH–positive blood cells were relatively high (85% and 80%), no clinical benefit was evident at the time of evaluation.
Parikh and colleagues8 reported UPD CN-LOH in chromosome 7, around the SBDS gene in hematopoietic cells from a patient with SDS carrying a compound, heterozygous, germline, pathogenic SBDS variant. Their finding is reminiscent of the current report of a CN-LOH replacement of a more damaging allele that acted as a null allele by the hypomorphic SBDS variant.
More recently, somatic genetic events in EIF6 (including interstitial chromosomal deletion, reciprocal translocation, and point mutations), that either sharply lowered eIF6 production or affected its function, have been shown to represent another type of SGR in the hematopoietic cells from patients with SDS (Tan S, Kermasson L, Hilcenko C, et al, manuscript submitted 2021).9
The findings of Lee et al further support the notion that several distinct somatic genetic events can modify the fate of the blood cells of patients with SDS, and likely impact their clinical evolution.
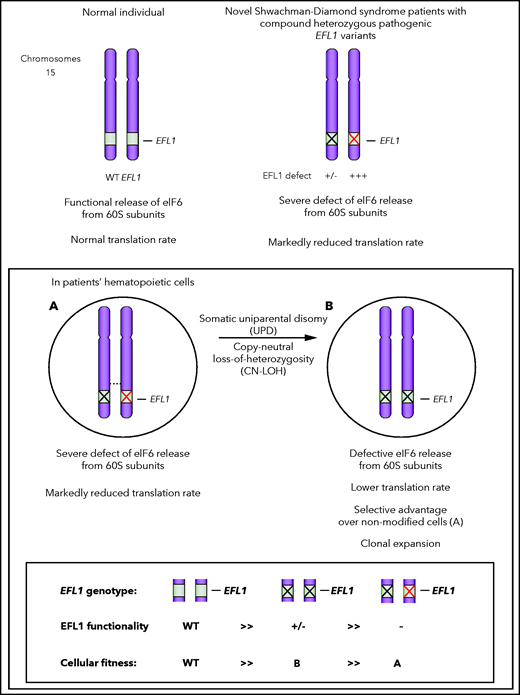
Segmental UPD of EFL1 in SDS: a schematic representation. Normal individual has 2 copies of wild-type (WT) EFL1 alleles (upper left). In the pathological situation (upper right), both EFL1 alleles carry deleterious/pathogenic variants (the X black allele is mildly defective, whereas the X red one is profoundly defective). Uniparental disomy (lower scheme) leads to the substitution of the most deleterious EFL1 allele (red X allele) by the less deleterious one (black X allele), contributing to mitigate the functional consequences of the most severe allele.
Segmental UPD of EFL1 in SDS: a schematic representation. Normal individual has 2 copies of wild-type (WT) EFL1 alleles (upper left). In the pathological situation (upper right), both EFL1 alleles carry deleterious/pathogenic variants (the X black allele is mildly defective, whereas the X red one is profoundly defective). Uniparental disomy (lower scheme) leads to the substitution of the most deleterious EFL1 allele (red X allele) by the less deleterious one (black X allele), contributing to mitigate the functional consequences of the most severe allele.
In the future, SGR will probably be detected in other conditions. Thus, one can imagine that cheaper and improved sensitive sequencing methods will enable the systematic search for or longitudinal follow-up of SGR events. This would represent considerable progress of patient-centered medicine by helping adopt the best therapeutic decisions for patients with Mendelian hematopoietic diseases, including SDS.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal