

Key Points
Distinct classes of SGK1 mutations identified in DLBCL converge on the production of stabilized SGK1 protein and AKT independence.
SGK1 should be considered as an oncogene and a potential therapeutic target in DLBCL.

Abstract
Serum and glucocorticoid-regulated kinase 1 (SGK1) is one of the most frequently mutated genes in diffuse large B-cell lymphoma (DLBCL). However, little is known about its function or the consequence of its mutation. The frequent finding of truncating mutations has led to the widespread assumption that these represent loss-of-function variants and, accordingly, that SGK1 must act as a tumor suppressor. In this study, instead, the most common SGK1 mutations led to production of aberrantly spliced messenger RNA neoisoforms in which translation is initiated from downstream methionines. The resulting N-terminal truncated protein isoforms showed increased expression related to the exclusion of an N-terminal degradation domain. However, they retained a functional kinase domain, the overexpression of which rendered cells resistant to AKT inhibition, in part because of increased phosphorylation of GSK3B. These findings challenge the prevailing assumption that SGK1 is a tumor-suppressor gene in DLBCL and provide the impetus to explore further the pharmacological inhibition of SGK1 as a therapeutic strategy for DLBCL.
Introduction
Serum and glucocorticoid-regulated kinase 1 (SGK1) has emerged as one of the most frequently mutated genes in DLBCL1,2 and also in lymphocyte-predominant Hodgkin lymphoma.3 Moreover, the presence of SGK1 mutation was the dominant genetic feature driving classification into the molecular subtypes termed “ST2” (for SGK1/TET2), “SGK1,” or “C4” in the 3 recent DLBCL genomic clustering studies.4-7 Despite the prominence of SGK1 mutations, little is known about its function or the molecular consequences of its mutation in B-cell lymphoma . However, the pattern of mutations, enriched for nonsense and splice variants, has led to the assumption that these represent loss-of-function mutations and that SGK1 must exert a tumor-suppressor function in DLBCL.4,8-10
SGK1 belongs to a family of serine and threonine kinases that includes AKT and is positively regulated by mTORC2 and PDK1.11 It acts as an AKT-independent effector of PI3K during cancer development12 and mediates resistance to AKT inhibition in epithelial cancers.13,14 Downstream targets of SGK1 are poorly defined, but there appears to be overlap with AKT.11 SGK1 is overexpressed in many solid-organ malignancies, including breast, prostate, and colon, where it is considered to function as an oncogene and is an attractive target for therapeutic inhibition.11,15 Given its apparent oncogenic function in epithelial malignancy, the question arises as to why loss-of-function mutation of SGK1 should be advantageous in B-cell lymphoma.
Study design
CRISPR-edited SGK1 mutant and control cell line clones were generated by electroporation of the PX458 plasmid (Addgene) encoding Cas9 and single guide RNA (gRNA) and a single-stranded 90-nucleotide DNA template containing the desired mutation and a SalI restriction site to facilitate screening. Correctly targeted clones were verified by Sanger sequencing. The resulting messenger RNA (mRNA) isoforms were identified using the 5' RACE System (rapid amplification of cDNA ends), version 2.0 (18374058; Invitrogen/Thermo Fisher Scientific), per the manufacturer’s instructions. Full experimental details are provided in the supplemental Methods, available on the Blood Web site.
Results and discussion
We pooled mutation data from 5 DLBCL sequencing studies.1,2,5,6,8 The results revealed widely scattered SGK1 mutations that included missense, nonsense, and splice variants, which predominantly affected the 5' region of the gene (Figure 1A; supplemental Table 1). This finding is consistent with SGK1 as a known target of AID-mediated aberrant somatic hypermutation.6,8 However, clear hotspots were evident, especially those affecting the splice regions either side of exon 2 (Figure 1A-B). The most frequent variants affected 2 adjacent nucleotides situated at the end of intron 1 and at the start of exon 2. These were identified in 35 and 49 patients, respectively. The exonic variant altered codon 26 (alanine); however, we hypothesized that these adjacent mutations may both affect mRNA splicing. We generated homozygous knockin clones by CRISPR editing of the intronic splice mutation into 2 lymphoma cell lines (Defauw and U2932; supplemental Figure 1A) and used 5' RACE to determine the resulting SGK1 transcript isoforms. In both cell lines, this method revealed the complete absence of the canonical transcript, which was replaced by 2 neoisoforms that had not been annotated (Figure 1C; supplemental Figure 1B). A longer neoisoform retained the entirety of intron 1. A shorter neoisoform excised intron 1 but used a cryptic splice site within exon 2, leading to loss of 14 nucleotides from the coding sequence. We then used publicly available RNA sequencing data to identify splice-mutant cases and looked for the presence of these aberrantly spliced SGK1 neoisoforms. We found evidence of both neoisoforms in cases with the intronic SGK1 splice mutant (Figure 1D). Furthermore, intron 1 retention was seen in cases with the adjacent exonic mutation (Figure 1D-E; supplemental Table 2). This confirms that the 2 most commonly identified SGK1 variants both disrupt mRNA splicing.
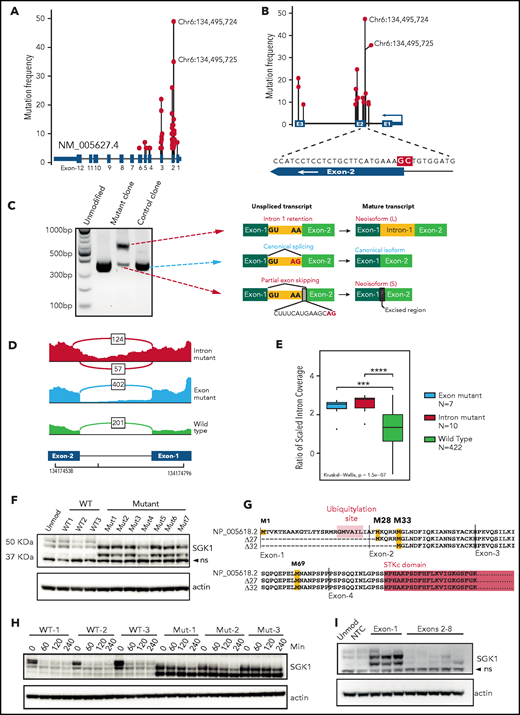
SGK1 mutations lead to aberrant splicing and hyperstable protein isoforms. (A) Distribution of mutations across the SGK1 locus from 5 published sequencing studies. The variant nucleotides depicted were identified in 5 or more cases. (B) Distribution of high-frequency variant nucleotides (identified in 10 or more cases) at mutation hotspots flanking exon 2. The magnified region shows the position of the 2 most frequent mutations highlighted in red. (C) Agarose gel electrophoresis showing polymerase chain reaction amplicons from 5' RACE performed on CRISPR-edited U2932 control and splice-mutant clones. The transcript neoisoform identified from Sanger sequencing of each band is shown. (D) Representative Sashimi plots from analysis of RNA sequencing data showing splicing across SGK1 intron 1. Data from a representative case are shown for each genotype. The number of reads corresponding to each splice junction is indicated. (E) An intron 1 retention score was calculated for each patient classified one of the genotypes. Significance was calculated using a Kruskal-Wallis test and pairwise comparisons with the Wilcoxon rank sum test. Adjusted P values: ***P < .001; ****P < .0001. (F) Immunoblot showing expression of SGK1 protein isoforms in multiple CRISPR-edited Defauw control and intronic splice mutant clones. The lower band is nonspecific (ns). (G) Predicted amino acid sequence from open reading frames identified in the WT and 2 aberrantly spliced neoisoforms. Predicted translational start sites are highlighted in yellow, the SGK1 kinase domain in pink, and the ubiquitination domain in orange. (H) Half-life study of SGK1 protein isoforms in CRISPR-edited control or splice-mutant clones showing immunoblot at the indicated times after addition of cycloheximide. (I) Immunoblot showing SGK1 protein isoform expression in Defauw-Cas9 cells transduced with single gRNAs targeting the indicated SGK1 exons. NTC, nontargeting control single gRNA.
SGK1 mutations lead to aberrant splicing and hyperstable protein isoforms. (A) Distribution of mutations across the SGK1 locus from 5 published sequencing studies. The variant nucleotides depicted were identified in 5 or more cases. (B) Distribution of high-frequency variant nucleotides (identified in 10 or more cases) at mutation hotspots flanking exon 2. The magnified region shows the position of the 2 most frequent mutations highlighted in red. (C) Agarose gel electrophoresis showing polymerase chain reaction amplicons from 5' RACE performed on CRISPR-edited U2932 control and splice-mutant clones. The transcript neoisoform identified from Sanger sequencing of each band is shown. (D) Representative Sashimi plots from analysis of RNA sequencing data showing splicing across SGK1 intron 1. Data from a representative case are shown for each genotype. The number of reads corresponding to each splice junction is indicated. (E) An intron 1 retention score was calculated for each patient classified one of the genotypes. Significance was calculated using a Kruskal-Wallis test and pairwise comparisons with the Wilcoxon rank sum test. Adjusted P values: ***P < .001; ****P < .0001. (F) Immunoblot showing expression of SGK1 protein isoforms in multiple CRISPR-edited Defauw control and intronic splice mutant clones. The lower band is nonspecific (ns). (G) Predicted amino acid sequence from open reading frames identified in the WT and 2 aberrantly spliced neoisoforms. Predicted translational start sites are highlighted in yellow, the SGK1 kinase domain in pink, and the ubiquitination domain in orange. (H) Half-life study of SGK1 protein isoforms in CRISPR-edited control or splice-mutant clones showing immunoblot at the indicated times after addition of cycloheximide. (I) Immunoblot showing SGK1 protein isoform expression in Defauw-Cas9 cells transduced with single gRNAs targeting the indicated SGK1 exons. NTC, nontargeting control single gRNA.
The sequence of both neoisoforms altered the reading frame and introduced premature stop codons. We were therefore surprised to see increased expression of a smaller protein isoform in edited clones when immunoblots were probed with a C-terminal–reactive SGK1 antibody (Figure 1F). Analysis of the nucleotide sequence of the 2 neoisoforms revealed open reading frames initiated from downstream methionines (M28, M33, and M69; Figure 1G). Taken together with the band sizes on immunoblot analysis, this finding suggests translation of protein isoforms that lack N-terminal 27 and 32 amino acids. These N-terminal truncations preserve the kinase domain but remove a ubiquitination site known to promote SGK1 degradation.16 We determined SGK1 protein half-life in control and splice-mutant edited clones and observed increased protein stability of both N-terminal truncated protein isoforms (40 minutes vs >4 hours; Figure 1H; supplemental Figure 1C). To establish whether other nonsplicing mutations of SGK1 may also result in the same effect we designed CRISPR gRNAs that targeted exons-1 to -7. All 3 gRNAs targeting exon 1 led to expression of stabilized, truncated protein isoforms that matched the size seen in the splice-mutant CRISPR clones. In contrast, gRNAs targeting downstream exons led to the expected depletion of the SGK1 protein (Figure 1I). This result suggests that, like the splice mutants, nonsense and frameshift variants within exon 1 result in translation from downstream methionines that exclude the degradation domain and thereby generate stabilized SGK1 protein isoforms. We conclude that multiple different classes of SGK1 mutation, frequently identified in DLBCL, converge on the production of hyperstable protein isoforms.
We examined growth of the SGK1 CRISPR-edited, splice-mutant clones using differential fluorescent labeling of control and mutant clones pooled in competitive coculture. This method revealed a growth advantage in splice-mutant clones over a 40-day time course (Figure 2A). We then used a recently described experimental culture system to clone the coding sequence of the wild-type (WT) and truncate SGK1 isoforms and express them in ex vivo cultured human germinal center B cells.17,18 Neither WT nor truncated SGK1 was associated with growth suppression and indeed both were associated with a small competitive advantage (Figure 2B). Taken together, the absence of growth suppression and the apparent competitive advantage in these 2 experimental systems are inconsistent with a tumor-suppressor function for SGK1 in B-cell lymphoma and support a prooncogenic effect instead.
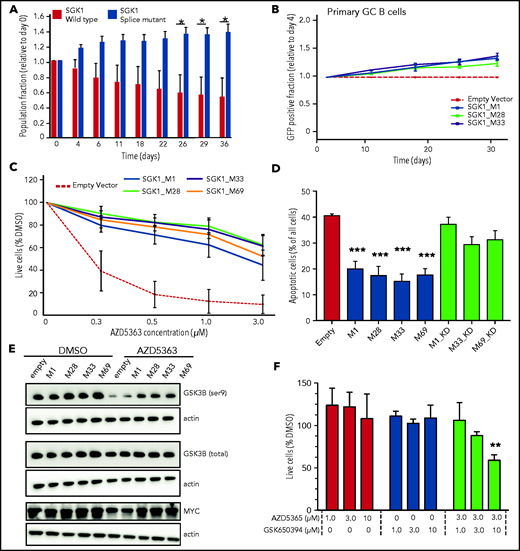
Increased SGK1 expression promotes cell growth and AKT independence. (A) CRISPR-edited control or splice-mutant Defauw clones were labeled with GFP and pooled for competitive growth. The relative proportion of control and mutant clones is shown over a 40-day time course. Data reflect 3 separate pairings of 3 WT and 3 mutant clones. Significance was calculated by paired Student t test. *P < .05. (B) Primary human germinal center B cells were immortalized with BCL6-2A-BCL2 and then transduced with cDNAs encoding canonical or truncated SGK1 isoforms. Constructs encoding SGK1 isoforms are named by the position of the initiating methionine. The frequency of cells transduced with each SGK1 isoform was quantified by flow cytometry at intervals of >30 days and normalized to day 4. (C) Cell viability measured by CellTiter-Glo after a 5-day treatment with the AKT inhibitor AZD5363 in BJAB cells transduced with control, canonical (M1) or N-terminal truncated (M28, M32, and M69) SGK1 isoforms. The figure shows an average of 4 independently conducted experiments; error bars represent standard error of the mean (SEM). (D) BJAB cells were transduced with the indicated canonical or truncated SGK1 isoforms, with or without the K127N kinase-dead (KD) mutation. Apoptosis was quantified by intracellular staining for caspase-3 and cleaved PARP 3 days after treatment with 500 nM AZD5363. Data show average and SEM of 3 separate experiments. Significance compared with the empty vector control was calculated by analysis of variance (ANOVA) and Dunnett’s multiple comparisons test. ***P < .001. (E) Immunoblot stained for phospho- and total GSK3B. Lysates are from BJAB cells stably transduced with the indicated canonical, WT (M1), or N-terminal truncated (M28, M32, and M69) SGK1 isoforms and treated with AZD5363 (500 nM) or dimethyl sulfoxide (DMSO) for 24 hours. Data are representative of 3 independent experiments. (F) Defauw cells were treated with the indicated concentration of AZD5363 and the SGK1 inhibitor GSK650394, either individually or in combination. Cell viability was measured by CellTiter-Glo. Data show average and SEM of 3 separate experiments. Significance compared with DMSO control was calculated by ANOVA and Dunnett’s multiple comparisons test. **P < .01.
Increased SGK1 expression promotes cell growth and AKT independence. (A) CRISPR-edited control or splice-mutant Defauw clones were labeled with GFP and pooled for competitive growth. The relative proportion of control and mutant clones is shown over a 40-day time course. Data reflect 3 separate pairings of 3 WT and 3 mutant clones. Significance was calculated by paired Student t test. *P < .05. (B) Primary human germinal center B cells were immortalized with BCL6-2A-BCL2 and then transduced with cDNAs encoding canonical or truncated SGK1 isoforms. Constructs encoding SGK1 isoforms are named by the position of the initiating methionine. The frequency of cells transduced with each SGK1 isoform was quantified by flow cytometry at intervals of >30 days and normalized to day 4. (C) Cell viability measured by CellTiter-Glo after a 5-day treatment with the AKT inhibitor AZD5363 in BJAB cells transduced with control, canonical (M1) or N-terminal truncated (M28, M32, and M69) SGK1 isoforms. The figure shows an average of 4 independently conducted experiments; error bars represent standard error of the mean (SEM). (D) BJAB cells were transduced with the indicated canonical or truncated SGK1 isoforms, with or without the K127N kinase-dead (KD) mutation. Apoptosis was quantified by intracellular staining for caspase-3 and cleaved PARP 3 days after treatment with 500 nM AZD5363. Data show average and SEM of 3 separate experiments. Significance compared with the empty vector control was calculated by analysis of variance (ANOVA) and Dunnett’s multiple comparisons test. ***P < .001. (E) Immunoblot stained for phospho- and total GSK3B. Lysates are from BJAB cells stably transduced with the indicated canonical, WT (M1), or N-terminal truncated (M28, M32, and M69) SGK1 isoforms and treated with AZD5363 (500 nM) or dimethyl sulfoxide (DMSO) for 24 hours. Data are representative of 3 independent experiments. (F) Defauw cells were treated with the indicated concentration of AZD5363 and the SGK1 inhibitor GSK650394, either individually or in combination. Cell viability was measured by CellTiter-Glo. Data show average and SEM of 3 separate experiments. Significance compared with DMSO control was calculated by ANOVA and Dunnett’s multiple comparisons test. **P < .01.
The concept of therapeutic targeting of SGK1 in lymphoma was previously proposed, either individually or in combination with AKT inhibition.3,19 However, it was previously unclear how this strategy may fit with apparent loss-of-function mutations of SGK1. Therefore, we examined the ability of SGK1 to act redundantly with AKT by overexpressing WT and truncated SGK1 neoisoforms in the lymphoma lines BJAB and SUDHL4, both of which lack expression of endogenous SGK1. Overexpression of either WT or truncated isoforms rendered cells resistant to the AKT inhibitor AZD5363. Truncated isoforms compensated for AKT at least as strongly as the WT isoform (Figure 2C-D; supplemental Figure 2A-C). The ability of canonical and truncated SGK1 isoforms to render cells resistant to AKT inhibition was dependent on the kinase activity of SGK1 isoforms. As such, resistance was abrogated either by the introduction of the SGK1 kinase–inactivating K127N mutation (Figure 2D) or by exposure to the SGK1 inhibitor GSK650394 (supplemental Figure 2D-E). To confirm pharmacological specificity, these findings were recapitulated with a different AKT inhibitor, MK2206 (supplemental Figure 2F). The physiological existence of SGK1 isoforms translated from downstream start sites of the canonical transcript has been noted previously, with the suggestion that shorter isoforms may preferentially phosphorylate a distinct set of downstream proteins, including GSK3B.20 GSK3B is known to be a direct target of both SGK1 and AKT.21 Immunoblots from transduced BJAB and SUDHL4 cells revealed that SGK1 maintained phosphorylation of GSK3B in the presence of AKT inhibition. This effect was more prominent in the presence of truncated SGSK1 isoforms (Figure 2E; supplemental Figure 2G) and abrogated by a K127N kinase–inactivating mutation of SGK1 (supplemental Figure 2H). SGK1-induced changes in GSK3B phosphorylation were associated with corresponding changes in expression of MYC, a known target of GSK3B (Figure 2E). Other targets of either canonical or truncated SGK1 isoforms in the setting of lymphomagenesis remain to be determined. However, the findings herein are evidence that stabilized, truncated SGK1 neoisoforms retain kinase activity, supporting the conclusion that many SGK1 mutations found in DLBCL lead to gain, not loss, of function. The findings also suggest that some AKT inhibitor-resistant lymphomas may be rendered sensitive by dual inhibition with AKT and SGK1 inhibitors. Indeed, SGK1 inhibition sensitized the SGK1-expressing cell line Defauw to AKT inhibition (Figure 2F).
We recognize that SGK1 has not emerged as a lymphoma-essential gene in recent lymphoma CRISPR screens.2,22 This fact may reflect redundancy with SGK3 or with other related kinases, such as AKT. Alternatively, CRISPR screening data may be complicated by the inclusion of gRNAs that target SGK1 exon 1, leading to increased protein expression. Finally, the oncogenic requirement of SGK1 may be restricted either to a specific stage of B cell differentiation or to a specific molecular subtype of DLBCL. Notably, there is currently no cell line model of the ST2 molecular subtype of DLBCL; therefore, the requirement for SGK1 has not been tested in the subtype in which its mutation is most frequently found. A lymphoma-promoting role for SGK1 in the ST2 molecular subtype is plausible, because SGK1 expression is known to be positively regulated by STAT3 activity.19 Indeed, both control and splice mutant clones showed elevated protein expression when STAT3 activation was induced with interleukin-21 (supplemental Figure 2I). Notably, JAK/STAT signaling is activated in the ST2 subtype by mutation of SOCS1 and DUSP2 and by activating mutation of STAT3 itself.4,5 Therefore, within the ST2 subtype, diverse genetic alterations may converge directly or indirectly on the upregulated expression of SGK1.
Overall, these results challenge the widely held assumption that SGK1 mutations lead to loss-of-function of a tumor suppressor in DLBCL. They reveal instead how deleterious and splice mutations lead to increased expression of truncated, hyperstable protein isoforms that retain kinase activity. Finally, they provide impetus to identify downstream targets of SGK1 and to explore the potential for SGK1 inhibition as therapy in specific subtypes of DLBCL .
Acknowledgments
The authors thank Jessica Bewick, Alice Mitchell, and Nicholaas Jonas (ENT Department, Addenbrooke’s Hospital, Cambridge, United Kingdom) for assistance in the collection of primary tonsil tissue; Kay Elston, Jane Price, and Joanna Baxter (Cambridge Blood and Stem Cell Bank) for collection and storage of primary tonsil tissue, and all members of the flow cytometry core for advice and support in flow cytometry.
This work was supported by the National Institute for Health Research (NIHR) Cambridge Bioresearch Center (BRC) Cell Phenotyping Hub and the NIHR Cambridge BRC (BRC-1215-20014) and by core funding from the UK Medical Research Council (MC_UU_12022/10) (S.S). D.H. was supported by a Clinician Scientist Fellowship and is currently supported by a Cancer Research UK (CRUK) Senior Research Fellowship (RCCFEL\100072). Research in the D.J.H. laboratory is supported by the Medical Research Council (MR/M008584/1) and the Kay Kendall Leukaemia Fund (KKL1144). The D.J.H. laboratory receives core funding from Wellcome (203151/Z/16/Z) and the Medical Research Council (MRC) to the Wellcome-MRC Cambridge Stem Cell Institute and from the CRUK Cambridge Centre (A25117).
The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Authorship
Contribution: J.G. and E.S. designed and conducted the experiments and analyzed the data; K.M. and Z.U. conducted the experiments; J.A.K. and I.W. analyzed the RNA sequencing data with supervision from S.A.S.; and D.J.H. obtained the funding, designed the experiments, analyzed the data, directed the research, and wrote the manuscript with contributions from J.G.
Conflict-of-interest disclosure: D.J.H. has received research funding from Gilead Sciences and Astra Zeneca. The remaining authors declare no competing financial interests.
Correspondence: Daniel J. Hodson, Wellcome-MRC Cambridge Stem Cell Institute, Jeffrey Cheah Biomedical Centre, Cambridge CB2 0AW, United Kingdom; e-mail: djh1002@cam.ac.uk.
Original data will be made available in response to an e-mail request to the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked ”advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal