

In this issue of Blood, 1 report that the most frequent mutations targeting the N-terminal region of serum and glucocorticoid-regulated kinase 1 (SGK1) in diffuse large B-cell lymphoma (DLBCL) have 1 common function: stabilization of the SGK1 protein. In this way, they solve the old conundrum of how seemingly inactivating mutations of seemingly oncogenic SGK1 promote DLBCL by identifying the true function of the mutations.
SGK1 belongs to a family of serine and threonine kinases, which also includes the known prosurvival AKT kinases. AKT kinases are frequently constitutively activated in germinal center B-cell–like (GCB) DLBCLs, most commonly by inactivation of the natural PI3K inhibitor PTEN.2 GCB-DLBCLs are highly sensitive to AKT and PI3K inhibitors but ultimately develop resistance. Although functions of AKT and SGK1 are not completely redundant, SGK1 can substitute for AKT to maintain survival and proliferation of malignant cells by acting as an AKT-independent effector of PI3K. In this way, SGK1 confers resistance to AKT inhibitors as demonstrated in DLBCL cell lines.3 SGK1 promotes oncogenesis and is highly expressed in different tumors, including ovary and breast carcinoma, gastric cancer, and prostate carcinoma.4
Similar to AKT, SGK1 is rarely mutated in tumors, with the exception of GCB-DLBCL and lymphocyte-predominant Hodgkin lymphoma.5,6 Moreover, SGK1 is one of the most frequently mutated genes in GCB-DLBCL.7 The mutation pattern of SGK1 is most likely the result of aberrant somatic hypermutation, which contributes to the development of lymphomas.8 Remarkably, SGK1 mutations differ from oncogenic mutations commonly observed in other kinases. They are not located in the kinase or regulatory domains, but rather target the N-terminus and comprise missense and nonsense variants, which are more typical for loss-of-function mutations.6
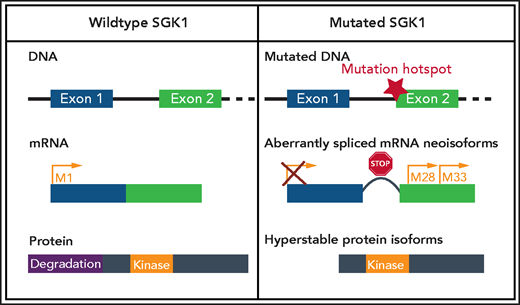
SGK1 mutations confer aberrant splicing and translation of hyperstable protein isoforms. The most common mutations occur in hotspot regions at the start of exon 2, which leads to the generation of 2 aberrantly spliced messenger RNA (mRNA ) neoisoforms. In addition, the mutations lead to disruption of the reading frame introducing premature stop codons. This provokes initiation of translation from downstream methionines, resulting in an N-terminally truncated hyperstable protein with preserved kinase domain but with lack of degradation domain. Created with BioRender.com.
SGK1 mutations confer aberrant splicing and translation of hyperstable protein isoforms. The most common mutations occur in hotspot regions at the start of exon 2, which leads to the generation of 2 aberrantly spliced messenger RNA (mRNA ) neoisoforms. In addition, the mutations lead to disruption of the reading frame introducing premature stop codons. This provokes initiation of translation from downstream methionines, resulting in an N-terminally truncated hyperstable protein with preserved kinase domain but with lack of degradation domain. Created with BioRender.com.
Given SGK1’s oncogenic function in epithelial malignancies and its ability to substitute AKT in terms of its prosurvival effect in DLBCL, Gao et al asked why apparent loss-of-function mutations of SGK1 should be advantageous in B-cell lymphoma. The authors reanalyzed the distribution of these mutations, noting the hotspots at the end of exon 1 and start of exon 2 and suggested that these mutations affect RNA splicing. With help of CRISPR/Cas9 editing, they homogenously introduced splice mutations into DLBCL cell lines and analyzed the resulting transcript isoforms. The canonical transcript disappeared and was replaced by aberrantly spliced neoisoforms, which were similar to those detected in DLBCL cases harboring intronic SGK1 mutations. Although the identified neoisoforms acquired altered reading frames containing premature stop codons, they were able to initiate translation of smaller proteins from downstream translation initiation codons in frame with the kinase domain. The transcribed protein did not contain the N-terminally located ubiquitination motif and, therefore, was distinguished by higher stability and intact kinase activity (see figure).
Of note, the idea of a protein-stabilizing effect of N-terminal SGK1 mutations is not new. Lu et al clearly demonstrated that the truncation of exon 1 increases SGK1 stability. In an attempt to identify the role of the frequent N-terminal mutations, A26V, A48V, and H51P mutations were introduced into the full-length SGK1 complementary DNA sequence and overexpressed in a human cell line. The mutations themselves did not increase the stability of the translated proteins, leaving open the question of their relevance.3
Thus, the work of Gao et al ultimately defines the hotspot mutations at the end of exon 1 and start of exon 2 in GCB-DLBCL as oncogenic. Future studies need to clarify whether they can serve as a reliable marker of low sensitivity to AKT inhibitors and whether the combination of SGK1 and AKT inhibitors will show a synergistic effect in tumors harboring these mutations. It is also of interest to elucidate the role of these mutations in lymphomagenesis. Experiments in transgenic animals harboring splice mutations of SGK1 should help to answer that question. In addition, this study highlights the complexity of mechanisms involved in oncogene activation and emphasizes the importance of CRISPR-mediated DNA editing of endogenous alleles to understand the meaning of mutations. It also exposes the rapidly widening gap between genomic studies, which identified a myriad of new mutations, and functional studies, which took years to unravel the impact of these mutations in oncogenesis.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal