

In this issue of Blood, describe an integrated analysis that includes genomic profiling, immunophenotyping, and outcome evaluation of one of the largest BCR-ABL1− B-lineage acute lymphoblastic leukemia (B-ALL) adolescent/adult patient cohorts treated within a single trial.1
Besides differentiating B-ALL from T-cell-ALL (T-ALL), with ALL being less frequent in both children and adults, a crucial discrimination in B-ALL lies in BCR-ABL1− and BCR-ABL1+ ALL. In BCR-ABL1+ ALL, a tyrosine-kinase inhibitor (eg, imatinib, dasatinib) is an essential element of therapy.2 However, most cases of B-ALL are BCR-ABL1− with only 2% to 5% of children and 20% to 30% of adults being BCR-ABL1+.3 BCR-ABL1− B-ALL is genetically very heterogeneous and is still insufficiently understood (compared with BCR-ABL1+ ALL). The era of genomics has influenced the World Health Organization (WHO) classification of BCR-ABL1− B-ALL. The 2016 updated version describes 7 distinct genomic categories and 2 provisional entities: B-ALL with iAMP21 and BCR-ABL1-like.4 BCR-ABL1–like ALL shares a similar gene expression profile with BCR-ABL1+ ALL, despite lacking the typical fusion gene occurring with the t(9;22), and has a poor outcome.5 If none of the genomic criteria for the 7 established and 2 provisional entities are met (ie, ALL’s lack of a unifying chromosomal alteration on cytogenetic analysis), patients are classified according to WHO as B-lymphoblastic leukemia/lymphoma not otherwise specified (NOS). This is the designation for a large number of adult patients with B-ALL. Pediatric ALL differs markedly from adult ALL with respect to cure rates (pediatric ALL 80% to 90% vs 40% to 50% in adults). To a significant extent, these differences are attributable to more high-risk genetic aberrations in adults.6 In pediatric ALL, much progress has been made in identifying novel genetic subgroups of B-ALL. Unfortunately, the genetic landscape of adult ALL is less well characterized. What are the challenges for classification of BCR-ABL− B-ALL in adults? Conventional cytogenetics are often unrevealing. Whereas cytogenetics and immunophenotyping are always used in diagnosing an adult with ALL, transcriptome sequencing and gene expression profiling are not routinely performed. However, with regard to the WHO classification and prognostication, it can be essential for genetically classifying B-ALL (eg, identifying BCR-ABL1–like ALL). Therefore, the study by Paietta et al is of great interest because it includes a cohort of 1229 BCR-ABL1− patients with B-ALL patients from 14 to 65 years of age (820 patients with ALL in a Medical Research Council (MRC) cohort, 409 in an ECOG-ACRIN cohort7) (see figure 1). Of those, 264 patients in the trial were comprehensively genetically analyzed. Of note, most patients were classified as B-ALL NOS, the subgroup we know the least about. In their study, Paietta et al identified several important findings about this subgroup. Certain immunophenotypic features correlated with distinct genetic subtypes, meaning that there was a clustering of antigen expressions in defined genotypes. In addition, mutations in TP53 were found to be enriched in low-hypodiploid patients with BCL2-Myc rearrangement, whereas IKZF1 alterations and mutations in the JAK-STAT pathway accumulated in BCR-ABL1-like ALL. Furthermore, an important focus of this study is how genetic subgroups and markers can be helpful for prognostication. For this purpose, this trial represents an ideal patient cohort, as all patients were uniformly treated without prior risk adaption. Of note, age and the white blood cell (WBC) count at the time of diagnosis are established parameters for risk stratification in adult ALL (and are still part of many adult trial protocols). In this trial, the protocol defined high-risk ALL factors as age >35 years, or a WBC count >30 × 109/L. In the past, the WBC count was also used for risk stratification in pediatric ALL. However, the improvement of genomic analysis and the identification of genetically defined prognostic groups has replaced the WBC count for use in risk stratification in many pediatric trial protocols. For instance, the Berlin-Frankfurt-Münster (BFM) pediatric ALL study group last used WBC count in the BFM 95 trial (recruitment completed in 2000). Applying the MRC/ECOG-ACRIN risk stratification based on WBC count and age as the stratifying criteria, 37% of patients were standard risk and 63% were assigned to the high-risk (pHR) group. Paietta et al correlated genetics and outcome in this large adult cohort and thus identified 3 molecular-risk groups: standard risk (5-year overall survival [OS], 53.2%), intermediate risk (5-year OS, 42%), and high risk (5-year OS, 12.9%). When the molecular risk classification was applied, 21.2% of the pHR were restratified to molecular standard risk and 18.2% of patients to molecular intermediate risk. This means, that approximately 40% of high-risk patients per protocol were not molecularly classified as high risk and had a superior outcome. One notable subgroup of patients was defined by DUX4 rearrangements with an excellent prognosis. These results demonstrate the direct clinical benefits of comprehensive genetic analysis. Another important observation was that antigen expression was not independently prognostic, but was significant only when used in conjunction with molecular markers.
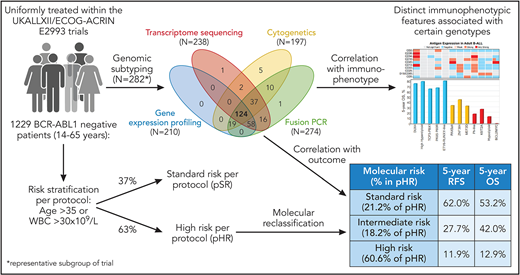
Design of an integrated analysis that includes genomic profiling, immunophenotyping, and outcome evaluation of patients with BCR-ABL1− B-ALL treated in the UKALLXII/ECOG-ACRIN E2993 trial.7 Of the patients previously classified in the protocol as high risk (based on WBC count and age), 39.4% were restratified as molecular standard risk (21.2%) or intermediate risk (18.2%). Professional illustration by Patrick Lane, ScEYEnce Studios.
Design of an integrated analysis that includes genomic profiling, immunophenotyping, and outcome evaluation of patients with BCR-ABL1− B-ALL treated in the UKALLXII/ECOG-ACRIN E2993 trial.7 Of the patients previously classified in the protocol as high risk (based on WBC count and age), 39.4% were restratified as molecular standard risk (21.2%) or intermediate risk (18.2%). Professional illustration by Patrick Lane, ScEYEnce Studios.
However, there are several questions that are of interest for future investigations. Minimal residual disease (MRD) monitoring, which has become a valuable standard tool for monitoring treatment success and risk stratification, was not performed in this trial.8 Therefore, we lack data combining comprehensive genetic profiling with MRD monitoring. In addition, an analysis of cooperating lesions between risk group–defining lesions was not included and leaves a critical question unanswered. Furthermore, the treatment landscape has changed for adult patients with B-ALL since the completion of the clinical trial. Novel substances (eg, blinatumomab, inotuzumab, and ozogamicin) and cellular therapies (eg, chimeric antigen receptor T-cell therapies) have been introduced in the treatment of B-ALL. In addition, the question of the role of allogeneic hematopoietic stem cell transplantation in defined genetic subgroups requires further analysis.9 It would be desirable for future clinical trials to implement genomic analysis at the time of diagnosis, to tailor treatment choices. Last, but not least, the high costs associated with such a comprehensive genetic analysis remain challenging for many health care systems. However, this study is an important step toward better understanding the heterogeneity of B-ALL and how this can be applied clinically.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal