

Key Points
del(17p) is an important prognostic factor in myeloma, even in the absence of TP53 mutation.
Isolated del(17p) should remain a high-risk feature in myeloma.

Abstract
Despite tremendous improvements in the outcome of patients with multiple myeloma in the past decade, high-risk patients have not benefited from the approval of novel drugs. The most important prognostic factor is the loss of parts of the short arm of chromosome 17, known as deletion 17p (del(17p)). A recent publication (on a small number of patients) suggested that these patients are at very high-risk only if del(17p) is associated with TP53 mutations, the so-called “double-hit” population. To validate this finding, we designed a much larger study on 121 patients presenting del(17p) in > 55% of their plasma cells, and homogeneously treated by an intensive approach. For these 121 patients, we performed deep next generation sequencing targeted on TP53. The outcome was then compared with a large control population (2505 patients lacking del(17p)). Our results confirmed that the “double hit” situation is the worst (median survival = 36 months), but that del(17p) alone also confers a poor outcome compared with the control cohort (median survival = 52.8 months vs 152.2 months, respectively). In conclusion, our study clearly confirms the extremely poor outcome of patients displaying “double hit," but also that del(17p) alone is still a very high-risk feature, confirming its value as a prognostic indicator for poor outcome.
Introduction
Despite huge therapeutic progress in multiple myeloma (MM), prognosis is still poor for high-risk patients. It is of great importance to identify these patients at diagnosis, because after decades of treatment adapted only to a patient’s age and renal function, the time has come to design risk-adapted treatment. Among all prognostic factors described for MM,1 genetic abnormalities displayed by tumor plasma cells play a major role.2,3 Deletion of the short arm of chromosome 17 (del(17p)) is a well-established high-risk feature in MM and is included in current disease staging criteria.4 However, several questions are still debated. The first concerns the prognostic threshold for clone size, with some investigators defining del(17p) if detected in only 1% of plasma cells.5 A recent meta-analysis of European data including more than 1000 patients with del(17p) clones of variable size confirmed previous data from the Intergroupe Francophone du Myélome (IFM),6 with a significant clinical impact of the subclonality value of 55% to 60% by FISH.7 With this threshold, del(17p) is observed in 8% of newly diagnosed MM (NDMM)6,8 and clearly impacts both progression free survival (PFS, median just over a year) and overall survival (OS, median 2-3 years).6,7 The second question addresses its molecular target; most studies have focused their analysis on the tumor suppressor gene TP53, localized in the minimal deleted region; however, the fact that the remaining allele is mutated in only a third of cases is surprising.9 A third question, related to the previous one, concerns the mono- or biallelic nature of the prognostic abnormality. Indeed, recent data suggest that only patients with a “double hit” biallelic inactivation of TP53 (del/mut most of the time) are at high-risk.10 In the study presented here, we demonstrate that an isolated del(17p) present in the majority of plasma cells, even if less unfavorable than a double hit, is still a prognostic indicator for poor outcome in MM.
Study design
The Toulouse Ethics Committee approved the study. Informed consent was obtained for all included patients. Clinical data were obtained from 121 NDMM patients displaying del(17p), treated by an intensive strategy, followed up for > 36 months, or having died of MM within 36 months post treatment, and for whom diagnosis was established between 2007 and 2017. We also established a control cohort of 2505 NDMM patients, defined by the absence of del(17p) (but including 307 patients (12.3%) displaying a translocation (4;14)), treated by intensive strategy, followed up for >36 months or having died of MM, and for whom diagnosis was established between 2007 and 2017. This control cohort was matched on age. All patients received either a doublet-based induction including bortezomib or a triplet-based including bortezomib and lenalidomide followed by ASCT and consolidation by the same regimen as induction, and some of them also received maintenance with lenalidomide (Table 1). Progression was determined based on criteria defined by the International Myeloma Working Group.11 Rates of PFS and OS were estimated by the Kaplan-Meier method. Tests were two-sided and P < .05 were considered significant. Statistical analyses were conducted using Prism.
Characteristics of patients with deletion 17p
| del(17p)/TP53wt | del(17p)/TP53mut | no del(17p) | |
|---|---|---|---|
| Patients, n | 76 | 45 | 2505 |
| Age at diagnosis, median (range), y | 61 (39-71) | 59 (40-71) | 58 (24-72) |
| Sex, n (%) | |||
| Male | 42 (55) | 25 (56) | 1451 (58) |
| Female | 34 (45) | 20 (44) | 1054 (42) |
| ISS, n (%) | |||
| 1 | 18 (29) | 7 (21) | 692 (32) |
| 2 | 28 (44) | 11 (32) | 950 (45) |
| 3 | 17 (27) | 16 (47) | 484 (23) |
| Patients, n | 63 | 34 | 2126 |
| Missing, n | 13 | 11 | 379 |
| del(17p), median (range), % | 76 (57-98) | 82 (58-96) | — |
| Treatment of multiple myeloma, n (%) | |||
| Doublet induction | 26 (34) | 17 (38) | 846 (34) |
| Triplet induction | 50 (66) | 28 (62) | 1659 (66) |
| Lenalidomide maintenance, n (%) | |||
| No | 66 (87) | 36 (80) | 2184 (87) |
| Yes | 10 (13) | 9 (20) | 321 (13) |
| Survival information | |||
| Deaths, n (%) | 39 (51) | 33 (73) | 601 (24) |
| Median follow-up (range), mo | 40.5 (1.8-148.4) | 36.0 (3.7-125.4) | 57.8 (1.6-185,2) |
| del(17p)/TP53wt | del(17p)/TP53mut | no del(17p) | |
|---|---|---|---|
| Patients, n | 76 | 45 | 2505 |
| Age at diagnosis, median (range), y | 61 (39-71) | 59 (40-71) | 58 (24-72) |
| Sex, n (%) | |||
| Male | 42 (55) | 25 (56) | 1451 (58) |
| Female | 34 (45) | 20 (44) | 1054 (42) |
| ISS, n (%) | |||
| 1 | 18 (29) | 7 (21) | 692 (32) |
| 2 | 28 (44) | 11 (32) | 950 (45) |
| 3 | 17 (27) | 16 (47) | 484 (23) |
| Patients, n | 63 | 34 | 2126 |
| Missing, n | 13 | 11 | 379 |
| del(17p), median (range), % | 76 (57-98) | 82 (58-96) | — |
| Treatment of multiple myeloma, n (%) | |||
| Doublet induction | 26 (34) | 17 (38) | 846 (34) |
| Triplet induction | 50 (66) | 28 (62) | 1659 (66) |
| Lenalidomide maintenance, n (%) | |||
| No | 66 (87) | 36 (80) | 2184 (87) |
| Yes | 10 (13) | 9 (20) | 321 (13) |
| Survival information | |||
| Deaths, n (%) | 39 (51) | 33 (73) | 601 (24) |
| Median follow-up (range), mo | 40.5 (1.8-148.4) | 36.0 (3.7-125.4) | 57.8 (1.6-185,2) |
ISS, International Staging System.
Bone marrow samples were obtained at diagnosis and shipped overnight to a central laboratory. Upon receipt, plasma cells were isolated using CD138+ MAC-Sorting (Miltenyi Biotec, Paris, France). Post-sorting purity was checked as previously described and only samples with ≥70% plasma cells after sorting were kept for the analysis. The mean purity was 96% and 93% of the samples displayed a purity ≥90%. Plasma cells were analyzed by FISH for t(4;14)(p16;q32) and del(17p) determination was by specific probe from Abbott Molecular (Paris, France) and Cytocell (Paris, France). Only del(17p) present in ≥55% plasma cells were taken into account.
For TP53 exonic mutations resequencing, a PCR amplicon based library was generated from multiplex PCR with 9 to 150ng genomic DNA input (11 specific couples of primers in supplemental Table, available on the Blood Web site). The library was sequenced with a MiSeq sequencer (Illumina, San Diego, USA) and Miseq Reagent kit V2 (paired-end sequencing 2 × 150 cycles). Sequencing depths from 100X to 47800X were obtained depending on samples or exons. Alignment was performed using BWA aligner and variant calling was performed using FreeBayes and Mutect2 variant callers. They were all verified on IGV.
Results and discussion
Characteristics of the patients are presented in Table 1.
Among 121 patients displaying del(17p), 45 (37.2%) also displayed a TP53 mutation (“double hit” MM). Of note, previous data show that virtually all patients with TP53 bi-allelic inactivation display the deletion in more than 55% of plasma cells.7 The OS of patients with TP53 biallelic inactivation was significantly shorter than patients with del(17p) alone (36.0 and 52.8 months, respectively, P = .004) (Figure 1A). Conversely, there was no statistically significant difference in PFS between the 2 groups (18.1 and 27.2 months, respectively, P = .1) (Figure 1B). Our data counteract an older study showing that 20 patients with TP53 biallelic inactivation had a similar prognosis to 34 patients with del(17p) alone.12 Conversely, in a dataset where both TP53 and del(17p) status were available, patients with TP53 biallelic inactivation (n = 7) had shorter PFS and OS than patients with monoallelic inactivation (n = 26, del(17p) or TP53 mutation).13
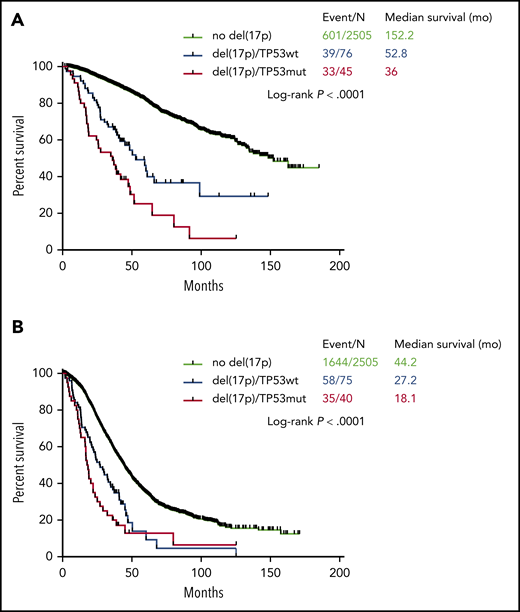
Kaplan-Meyer survival of myeloma patients according to del(17p). (A) Overall survival. (B) Progression-free survival. The blue curve corresponds to patients with del(17p) alone, the red curve to patients with biallelic inactivation of TP53, and the black curve to patients without del(17p) (control cohort).
Kaplan-Meyer survival of myeloma patients according to del(17p). (A) Overall survival. (B) Progression-free survival. The blue curve corresponds to patients with del(17p) alone, the red curve to patients with biallelic inactivation of TP53, and the black curve to patients without del(17p) (control cohort).
In this study, we chose a variant allelic fraction (VAF) cutoff of 0.4. Seven patients with del(17p) displayed a TP53 mutation with a VAF ranging from 0.1 to 0.4; when transferred in the TP53 biallelic inactivation group, the difference in PFS and OS between the 2 groups was similar (data not shown).
In order to determine if del(17p) alone can be considered a poor prognostic factor in MM, we compared the 76 patients with del(17p) alone to the 2505 patients without del(17p). We observed that PFS and OS were significantly shorter for patients with isolated del(17p) (PFS 27.2 versus 44.2 months, P < .0001; OS 52.8 versus 152.2 months, P < .0001 (Figure 1).
One unresolved question is the prognostic role of isolated TP53 mutations. Our control cohort was based on the absence of del(17p) and not on TP53 status, which means that patients with TP53 mutation only were included. It remains to be determined in large cohorts if monoallelic mutation of TP53 is a poor prognostic feature in MM.9 If the answer is yes, this mutation should be systematically sought in clinical practice. The NGS approach for routine risk assessment has the advantage of being informative on copy number abnormalities, translocations of interest such as t(4;14) and TP53 mutations in one single technique.14 Of note, 73% of the patients with del(17p) alone and 52% of those with TP53 biallelic inactivation were not International Staging System (ISS)-3 (Table 1), which means that they were not classified in the Revised-ISS-3 subgroup.4 Given the current number of available drugs, all patients with del(17p) in most of their plasma cells should benefit from the most powerful combinations as in other cancers. If evidence suggest that recent developments in immunotherapy such as CAR-T cells or T cell engagers are of particular interest to these patients, these approaches should be used in earlier lines. Of note, both del(17p) and TP53 mutations can be acquired during evolution, meaning that both analyses should be performed again at the time of any relapse.
In summary, we demonstrate that an isolated del(17p) in more than 55% of plasma cells, even if less unfavorable than a biallelic inactivation of TP53, is still a poor prognosis factor in MM, and should remain the major factor to define high risk patients.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
No additional data will be made available by the authors.
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by National Institutes of Health, National Cancer Institute grants PO1-155258 and P50-100707 (H.A.-L.) and the Cancer Pharmacology of Toulouse and Region (CAPTOR) program. The Centre de Recherches en Cancérologie de Toulouse Team 13 is supported by the Fondation ARC (Association pour la Recherche sur la Cancer, grant PGA1*20160203788).
Authorship
Contribution: J.C., A.P., L.B., and H.A.-L. designed the research and analyzed data, J.C. and H.A.-L. wrote the manuscript, which was reviewed and edited by the other coauthors; J.C. performed the statistical analysis; and all other authors provided study samples and clinical data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hervé Avet-Loiseau, Unit for Genomics in Myeloma, Institut Universitaire du Cancer – Oncopole, 1 Av Irène Joliot-Curie, 31100 Toulouse, France; e-mail: avetloiseau.herve@iuct-oncopole.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal