

Key Points
Whole-exome sequencing of twins with ALL identified the GNAO1 R209C mutation as a novel second hit for ETV6-RUNX1+preleukemia.
Reciprocal activation of ETV-6-RUNX1 fusion and GNAO1 R209C mutation induces leukemogenesis by activating PI3K/Akt/mTOR signaling.

Abstract
Leukemogenesis is characterized by chromosomal rearrangements with additional molecular disruptions, yet the cooperative mechanisms are still unclear. Using whole-exome sequencing of a pair of monozygotic twins who were discordant for childhood acute lymphoblastic leukemia (ALL) with ETV6-RUNX1 (E/R) gene fusion successively after birth, we identified the R209C mutation of G protein subunit α o1 (GNAO1) as a new ALL risk loci. Moreover, GNAO1 missense mutations are recurrent in ALL patients and are associated with E/R fusion. Ectopic expression of the GNAO1 R209C mutant increased its GTPase activity and promoted cell proliferation and cell neoplastic transformation. Combined with the E/R fusion, the GNAO1 R209C mutation promoted leukemogenesis through activating PI3K/Akt/mTOR signaling. Reciprocally, activated mTORC1 phosphorylated p300 acetyltransferase, which acetylated E/R and thereby enhanced the E/R transcriptional activity of GNAO1 R209C. Thus, our study provides clinical evidence of the functional cooperation of GNAO1 mutations and E/R fusion, suggesting GNAO1 as a therapeutic target in human leukemia.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common malignant pediatric cancer, and, despite cure rates exceeding 90% in children, it remains a major cause of mortality in children and adults.1,2 The t(12;21) ETV6 (TEL)-RUNX1 (AML1)(E/R) gene fusion is one of the most common chromosomal translocations in childhood ALL,3,4 but E/R gene fusion alone may not be sufficient to drive full-blown disease.2,5,6 However, the cooperative mechanisms remain elusive.
Heterotrimeric G proteins are molecular switches that control signal transduction, and identification of their oncogenic mutations has provided insights into the molecular mechanisms that induce tumorigenesis.7-11 G protein subunit α o1 (GNAO1, also called Gαo) is the α subunit of Go.12 Mutations in Gα proteins have been identified in human tumors and promote malignant transformation and tumorigenesis by rendering the G proteins constitutively active in the GTP-bound conformation, such as GNAS (Gαs) in pituitary adenomas13,14 and GNAQ (Gαq) and GNA11 in uveal melanoma.7,8 Although mutations in GNAO1 have been found to be related to early infantile epileptic encephalopathy and movement disorder15-17 and have been described to be oncogenic when artificially introduced into cultured cells,18-20 no such mutations in GNAO1 have been functionally characterized in human cancers.
In this study, using whole-exome sequencing of a pair of monozygotic twins who were discordant for E/R+ childhood leukemia, we identified GNAO1 R209C mutation as a driver in leukemia. We then used cell culture and orthotopic xenograft models to assess the role of GNAO1 R209C mutation in the proliferation, colony formation, and tumor growth of leukemia cells. Finally, we determined the cooperative mechanisms by which GNAO1 R209C mutation and E/R gene fusion promote leukemogenesis.
Materials and methods
Patient samples
A pair of monozygotic twins was diagnosed with leukemia within 110 days after birth. Peripheral blood mononuclear cells (PMBCs) and bone marrow (BM) samples were collected in Shanghai Children’s Medical Center (SCMC) at different stages: twin A, at onset and at complete remission, and twin B, before onset and at onset. BM samples of another 102 patients with acute myeloid leukemia (AML) and 106 with ALL were also collected at SCMC. The study was approved and supervised by the SCMC Ethics Committee, according to the Declaration of Helsinki. All subjects provided written consent for banking of tissue and future research use of the samples, in accordance with the regulations of the institutional review board of SCMC.
Whole-exome sequencing
Genomic DNAs were extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Whole-exome capture libraries were prepared according to standard protocols with the SureSelect Human All Exon 50-Mb Kit and assessed with the 2100 Bioanalyzer (both from Agilent Technologies, Palo Alto, CA) with 150-bp paired-end reads. Exome data processing, variant calling, and variant annotation were performed as previously described.21 Genetic mutations reported in this study were further validated by Sanger sequencing.
Cell culture
HEK-293T, leukemia Reh, UoCB6, Nalm6, K562, and TPH1 cells were from American Type Culture Collection (ATCC; Manassas, VA). Leukemia cells were cultured in 10% fetal bovine serum/RPMI-1640 medium. HEK-293T cells were cultured in 10% fetal bovine serum+Dulbecco’s modified Eagle’s medium. All cell lines in this study were authenticated by using STR DNA fingerprinting by Shanghai Biowing Applied Biotechnology Co, Ltd (Shanghai, China), and mycoplasma infection was detected with the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich).
Plasmids, lentivirus production, and infection
Human ETV-6-RUNX1 and GNAO1 coding regions were cloned into the pLVX-Puro Vector (Clontech) with HA, Flag tag, or GFP. (His)6-tagged GNAO1 was constructed with a pcDNA3 vector (Clontech). Point mutations were constructed using site-directed mutagenesis and confirmed by DNA sequencing. The lentiviral constructs were transfected by packaging plasmids into HEK293T cells using the calcium phosphate method to produce a replication-defective virus. The supernatant was harvested 48 h later and concentrated, and cells were virally transduced with 8 μg/mL polybrene (Sigma-Aldrich).
Purification of recombinant proteins
(His)6-tagged GNAO1 WT and R209C mutant proteins were purified from HEK293T cells on a Ni+-NTA column at 4°C. The purified recombinant GNAO1 proteins were examined by Coomassie brilliant blue staining and western blot analysis. The aliquots were stored at 80°C until use.
GTPase activity analysis
GTPase activity analysis was performed with a GTPase colorimetric assay kit (Innova Biosciences) according to the manufacturer’s instruction. In brief, purified GNAO1 WT and R209C mutant proteins were incubated with substrate/buffer mixture (50 mM Tris-HCl [pH 7.5], 2.5 mM MgCl2, and 0.5 mM GTP) at 25°C, and the reactions were stopped by PiColorLock Gold Mix. The released free phosphate was quantified at a wavelength in the range of 590 to 660 nm.
Structural analysis
Simulated crystal structural analysis of GNAO1 R209C mutant was performed with PyMol (www.pymol.org).
Immunoprecipitation and western blot analysis
Cells were lysed in a buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 2 mM Na3VO4, 5 mM NaF, 1% Triton X-100 and protease inhibitor cocktail) at 4°C for 30 minutes. The lysates were centrifuged, and the protein concentrations were determined. Equal amounts of cell lysates were loaded. Immunoprecipitation (IP) and western blot analysis (WB) were performed as previously described22 with antibodies against β-actin (I-19), Src (SRC 2; Santa Cruz Biotechnology); HA (BD Transduction Laboratories); Flag (MS2; Sigma-Aldrich); phospho-p44/42 MAPK (Thr202/Tyr204; 9101), p44/42 MAPK (9102), phospho-Akt (Ser473; 4060), Akt (9272), STAT-3 (124H6; 9139), phospho-Src (TYR416; 6943), S6 (5G10; 2217), phospho-S6 (Der235/236; 4858), acetylated-lysine (9441), p300 (86377), TAF1 (12781), and phospho-STAT3 (Tyr705; D3A7) (all from Cell Signaling Technology); and GNAO-1 (12635-1-AP; Proteintech).
RT-PCR and qRT-PCR assays
Reverse transcription-polymerase chain reaction (RT-PCR) was performed as previously described.22 Quantitative RT-PCR (qRT-PCR) was performed with the QuantiTect SYBR Green PCR Kit (Qiagen, Valencia, CA) on a Rotor-Gene 6000 series PCR cycler (Corbett Research, Valencia, CA). All messenger RNA quantification data were normalized to an internal control, ACTB. Primers were as follows: GNAO1: 5′-TGTGTGATGTGGTGAGTCGG-3′ and 5′-ATCCAGGCTGTCCAGGTAGT-3′; ACTB: 5′-CATGTACGTTGCTATCCAGGC-3′ and 5′-CTCCTT AATGTCACGCACGAT-3′; and ETV6-RUNX1: 5′-CTCTGTCTCCCCGCCTGAA-3′ in ETV6 and 5′-CGGCTCGTGCTGGCAT-3′ in RUNX1.
Cell proliferation analysis
Cell proliferation analysis was performed with a Click-iT EdU Assay Kit (Thermo Fisher Scientific).
Colony formation analysis
A soft agar colony-formation assay was performed as we previously described.22 In brief, various cells were seeded in a top layer of 0.4% Noble Agar with a bottom layer of 0.8% Noble Agar in each of the triplicate wells of a 24-well plate. Colonies were scored after 2 to 3 weeks, and data were analyzed using GraphPad Software.
Flow cytometry analysis
Flow cytometry was performed as previously described.23
Tumorigenesis studies
White severe combined immunodeficiency (SCID) female mice aged 6 to 8 weeks (SLAC, Shanghai, China) were used. The mice were randomly divided into 5 per group. In total, 1 × 106 leukemia cells were injected into recipients through the tail vein, as previously described.24 Mice were euthanized when ALL symptoms developed. All animal experiments were performed in accordance with a protocol approved by Shanghai Jiao Tong University Institutional Animal Care and Use Committee.
Chromatin Immunoprecipitation-qPCR
Chromatin immunoprecipitation (ChIP) was performed with a kit (Millipore-Upstate). Immunoprecipitated DNA was purified after phenol extraction and used for qPCR. Primers for GANO1 (promoter) were 5′-AGTGCAAAGACGGCCTCCACC-3′ and 5′-GCCAGGGTGCCCGGAGTGG-3′.
Promoter reporter and dual luciferase assays
The GNAO1 promoter was amplified by PCR with the primers 5′-TATACCTAAGGTCAATGATAGT-3′ and 5′- CGCGAGTGCGCCAGCGGCTCTG-3′ and then inserted into a pGL3.0-Basic vector. For normalization of transfection efficiency, the pRL-TK (Renilla luciferase) reporter plasmid was added to each transfection. Luciferase activity was quantified with a Dual-Specific Luciferase Assay Kit (E1910; Promega).
Statistics
Statistical analyses were performed with Prism 5.0 software (GraphPad). P < .05 was considered statistically significant.
Results
Mutational analysis in a monozygotic twin pair identified the GNAO1 R209C mutation as a second hit for ETV6-RUNX1+ preleukemia
To define the cooperative mechanisms that are associated with primary E/R gene fusion during the development of leukemia, specifically in the clinic, we focused on 1 pair of monozygotic twins who were diagnosed with leukemia within 110 days of birth (Figure 1A). To characterize the putative fusion genes that resulted in leukemia and detect all potential cooperative somatic genomic changes, we collected PBMCs and 4 BM samples from the twins (from twin A, samples were collected at onset of leukemia and at complete remission; from twin B, the samples were collected before onset and at onset (Figure 1A). We then performed whole-exome sequencing and found an intrachromosomal translocation that gave rise to the ETV6-RUNX1 (E/R) fusion gene (Figure 1B). This fusion gene is also known as TEL/AML1, the most frequent gene fusion in childhood ALL and a crucial factor for initiation of disease.1,3,25

Mutational analysis in a monozygotic twin pair identified the GNAO1 R209C mutation as a second hit for ETV6-RUNX1+ preleukemia. (A) Schematics of a pair of monozygotic twins who were diagnosed with leukemia within 110 days after birth. (B) Schematics of the t(12; 21) ETV6-RUNX1 fusion gene and validation by PCR sequencing. (C) Sanger sequencing of the twins’ samples. GNAO1 c.625C>T point mutations in the twins’ samples at onset of leukemia are indicated: twin A, at the onset of leukemia and at remission; twin B, before the onset of leukemia and at the onset of leukemia. (D) Allele frequency analysis of gene mutations by deep genomic sequencing in the onset specimens from the twins, corresponding to supplemental Table 1. (E) Allele frequency analysis of the ETV6-RUNX1 fusion gene and GNAO1 R209C mutation in twin B before the onset of leukemia.
Mutational analysis in a monozygotic twin pair identified the GNAO1 R209C mutation as a second hit for ETV6-RUNX1+ preleukemia. (A) Schematics of a pair of monozygotic twins who were diagnosed with leukemia within 110 days after birth. (B) Schematics of the t(12; 21) ETV6-RUNX1 fusion gene and validation by PCR sequencing. (C) Sanger sequencing of the twins’ samples. GNAO1 c.625C>T point mutations in the twins’ samples at onset of leukemia are indicated: twin A, at the onset of leukemia and at remission; twin B, before the onset of leukemia and at the onset of leukemia. (D) Allele frequency analysis of gene mutations by deep genomic sequencing in the onset specimens from the twins, corresponding to supplemental Table 1. (E) Allele frequency analysis of the ETV6-RUNX1 fusion gene and GNAO1 R209C mutation in twin B before the onset of leukemia.
In the search for putative cooperative somatic genomic alterations in the twins, we identified shared a missense c.625C>T point mutation in GNAO1 located on chromosome 16 (Figure 1C; supplemental Table 1, available on the Blood Web site), leading to a p.Arg209Cys (R209C) mutation of the GNAO1 protein, which is also a mutation hotspot in pediatric epilepsies.26-28 Analysis of sequencing reads and further genotyping using a Sequenom assay showed an ∼50% allele frequency for the GNAO1 c.625C>T point mutation in both twins (Figure 1D; supplemental Table 1), suggesting that the point mutation was present in almost all leukemic cells from both twins. Because the separate occurrence of the same mutations in twins is rare, our results suggest that the twins may have obtained the same leukemic colony by sharing blood cells during the fetal stage.
Given the previously documented role of GNAO1 protein–mediated neoplastic transformation,10,19,20 it was of great interest to us to explore whether the GNAO-1 R209C mutation could be a cooperative event in the development of E/R+ leukemia. We performed PCR sequencing using HiSeq 2000 of the BM sample from twin B (at 50 days old) before the onset of leukemia and obtained 0.3 million reads per sample. Allele frequency analysis showed that the frequency of occurrence of the E/R gene fusion was 12.5%, and the frequency of GNAO1 R209C was only 0.35% (Figure 1E). The significant difference indicates that the prenatal first hit generated E/R+ preleukemia cells, and the second hit generated GNAO1 R209C–mutated leukemia cells in 1 fetus. Then, the leukemia cells expanded in utero and thus became shared between the twins. Moreover, cells with the GNAO1 R209C mutation rapidly proliferated in a 2-month period and became the major clone at the onset of the disease (Figure 1D). These data suggest that the GNAO1 R209C mutation is a newly identified leukemia driver.
GNAO1 missense mutations in patients with ALL
Because GNAO1 point mutations, such as G184S, G203R,15,16 R243H,20 and Q205L,18 are important in the development of cancer and diseases (Figure 2A), we analyzed whole-exome sequencing data for 102 AML and 106 ALL samples to examine the prevalence of GNAO1 mutations in a general population of patients with leukemia. We identified only 3 novel somatic GNAO1 mutations in patients with ALL (Figure 2A; supplemental Table 2). Two of the 3 mutations harbored an E/R fusion (supplemental Table 2), which was validated by Sanger sequencing (supplemental Figure 1), and another patient with a GNAO1 mutation also had an IGH/IGK mutation (supplemental Table 2). A high frequency of each GNAO1 mutation was revealed by deep genomic sequencing (Figure 2B). We further searched the putative GNAO1 mutations in the Catalogue of Somatic Mutations in Cancer (https://cancer.sanger.ac.uk/cosmic) and found a K317K mutation in a patient with AML and a T329M mutation in a patient with acute leukemia of ambiguous lineage (Figure 2A; supplemental Table 3). These data demonstrate that GNAO1 mutations are recurrent in human leukemia and may be associated with E/R or other driver mutations in leukemogenesis.
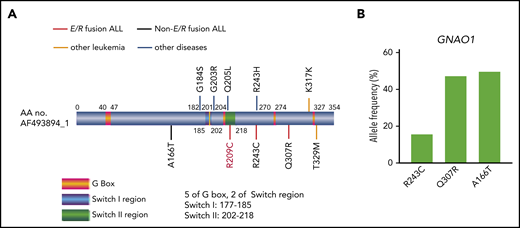
GNAO1 mutations in patients with ALL or other diseases. (A) The type and position of each GNAO1 mutation with or without the ETV6-RUNX1 (E/V) fusion identified are shown. R209C, R243C, and Q307R mutations in a patient with E/R fusion ALL, A166T mutation in a patient with non-E/R fusion ALL, T329M mutation in a patient with acute leukemia of ambiguous lineage, K317K mutation in a patient with AML, G184S and G203R mutation in patients with early infantile epileptic encephalopathy,15,16 R243H mutation in a patient with breast cancer,20 and Q205L mutation with NIH-3T3 transformation.18 (B) Allele frequency analysis of new GNAO1 mutations by deep genomic sequencing in the specimens identified, corresponding to supplemental Table 2.
GNAO1 mutations in patients with ALL or other diseases. (A) The type and position of each GNAO1 mutation with or without the ETV6-RUNX1 (E/V) fusion identified are shown. R209C, R243C, and Q307R mutations in a patient with E/R fusion ALL, A166T mutation in a patient with non-E/R fusion ALL, T329M mutation in a patient with acute leukemia of ambiguous lineage, K317K mutation in a patient with AML, G184S and G203R mutation in patients with early infantile epileptic encephalopathy,15,16 R243H mutation in a patient with breast cancer,20 and Q205L mutation with NIH-3T3 transformation.18 (B) Allele frequency analysis of new GNAO1 mutations by deep genomic sequencing in the specimens identified, corresponding to supplemental Table 2.
The GNAO1 R209C mutation increases its enzymatic activity and promotes cell proliferation and neoplastic transformation
To demonstrate the mechanism by which the GNAO1 R209C mutation causes childhood ALL, we evaluated its impact on GNAO1 enzymatic activity. Using Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) analysis, we found that the R209 residue of GNAO1 is located in a switch II domain (supplemental Figure 2A), a region important for guanine nucleotide-dependent regulation of downstream effectors such as PLCβ.26 The GNAO1 R209 residue is evolutionarily conserved from fish to humans (supplemental Figure 2B) and is also highly conserved in the Gα protein family (supplemental Figure 2C), suggesting that the residue is important in the function of G proteins. Then, we purified the GNAO1 wild-type (WT) and R209C mutant and analyzed their enzymatic activity with a GTPase colorimetric assay kit. Compared with WT GNAO1 activity, the activity of the R209C mutant was markedly higher (Figure 3A). A simulated crystal structure of the GNAO1 R209C mutant was made using PyMol (www.pymol.org) (Figure 3B). The side chain of R209 forms a salt bridge with that of Glu246 (E246), which has been shown to be important for GTP-bound active Gα-containing complexes.26 The R209C mutation disrupted their interaction, thereby destabilizing the Gα-containing complexes in GTP-bound active states. Thus, these data suggest that the GNAO1 R209C mutant is constitutively active by accelerated nucleotide exchange.
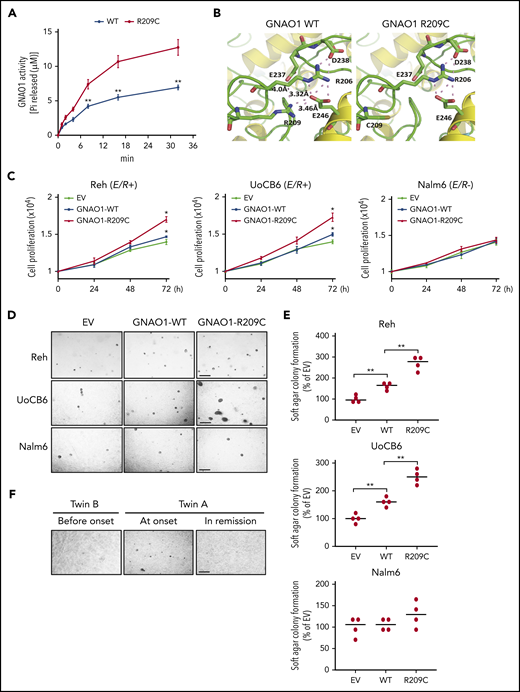
GNAO1 R209C mutation increases its enzymatic activity and promotes cell proliferation and neoplastic transformation. (A) Enzymatic activity analysis of WT GNAO1 and the R209C mutant. A GTPase activity analysis was performed with the GTPase colorimetric assay kit. (B) The simulated crystal structures of WT GNAO1 and the R209C mutant. (C) Effects of ectopic expression of WT GNAO1 or the R209C mutation on cell proliferation in Reh, UoCB6, and Nalm6 cells. Cell proliferation analysis was performed using the Click-iT EdU Assay Kit. (D) Representative images of soft agar colony formation. Scale bar, 1 mm. (E) Quantification of soft agar colony formation in panel D. (F) Representative images of soft agar colony formation of the PBMC specimens from twin B before the onset of leukemia, twin A at the onset of leukemia, or twin A at remission. Data are representative of 3 or 4 independent experiments with similar results. Scale bar, 1 mm. (A,C,E) data are expressed as the mean ± SD. *P < .05; **P < .01, by 2-tailed Student t test.
GNAO1 R209C mutation increases its enzymatic activity and promotes cell proliferation and neoplastic transformation. (A) Enzymatic activity analysis of WT GNAO1 and the R209C mutant. A GTPase activity analysis was performed with the GTPase colorimetric assay kit. (B) The simulated crystal structures of WT GNAO1 and the R209C mutant. (C) Effects of ectopic expression of WT GNAO1 or the R209C mutation on cell proliferation in Reh, UoCB6, and Nalm6 cells. Cell proliferation analysis was performed using the Click-iT EdU Assay Kit. (D) Representative images of soft agar colony formation. Scale bar, 1 mm. (E) Quantification of soft agar colony formation in panel D. (F) Representative images of soft agar colony formation of the PBMC specimens from twin B before the onset of leukemia, twin A at the onset of leukemia, or twin A at remission. Data are representative of 3 or 4 independent experiments with similar results. Scale bar, 1 mm. (A,C,E) data are expressed as the mean ± SD. *P < .05; **P < .01, by 2-tailed Student t test.
To determine the functions of the GNAO1 R209C mutant, we detected the effects of ectopic expression of the GNAO1 R209C mutant on cell proliferation in ALL E/R-positive Reh and UoCB6 cells24,29,30 and E/R-negative Nalm631 cells (supplemental Figure 3). Compared with the empty vector (EV) control, the ectopic expression of GNAO1 WT significantly promoted cell proliferation (Figure 3C) and soft agar colony formation (Figure 3D-E) of Reh and UoCB6 cells but not of Nalm6 cells. Compared with overexpression of the WT, overexpression of the GNAO1 R209C mutant further enhanced cell proliferation (Figure 3C) and soft agar colony formation (Figure 3D-E) of the Reh and UoCB6 cells. To further validate this observation, we collected PBMCs from twin B before the onset of leukemia, from twin A at the onset of leukemia, and from twin A at remission and performed a soft agar colony formation analysis. Only PBMCs from twin A at the onset formed colonies (Figure 3F). These results support that the GNAO1 mutation induces neoplastic transformation in combination with the E/R fusion.
The GNAO1 R209C mutation promotes ETV6-RUNX1+ leukemogenesis
To further determine the effects of the GNAO1 R209C mutation in vivo, we used an orthotopic ALL xenograft model in immunodeficient mice. Reh cells with stable expression of WT GNAO1, the R209C mutation, or a GFP control were injected IV into the mice through the tail vein. Compared with the GFP control, WT GNAO1 markedly increased spleen size and weight (Figure 4A-B) and enhanced BM infiltration (Figure 4C-D). As expected, these effects were more pronounced in mice with the GNAO1 R209C mutant than in those with WT GNAO1 or the GFP control (Figure 4A-D). In addition, Kaplan-Meier analysis showed that the animals with the R209C mutant had the poorest prognosis (Figure 4E). We also assessed the effects of overexpression of WT GNAO1 and the R209C mutant in Nalm6 cells with non-E/R gene fusion and K562 cells with BCR/ABL gene fusion. However, there was no significant difference in BM infiltration (supplemental Figures 4A-B and 5A-B) or spleen weight (supplemental Figures 4C and 5C). Taken together, these data support the notion that GNAO1 activation induces ALL tumorigenesis with E/R cooperation.

GNAO1 R209C mutation promotes ETV6-RUNX1+ leukemogenesis. (A) Representative images of the effects of ectopic expression of GNAO1 WT or R209C mutant on mouse spleen size at days 32 to 35 after injection. Reh cells expressing WT GNAO1, the R209C mutation, or a GFP control (GFP+) were injected into recipients through the tail vein. GFP−, negative GFP cell control. Images represent the results of 5 mice per group of 3 independent experiments. Scale bars, 1 cm. (B) Quantification of spleen weight in panel A. (C) Flow cytometry analysis of BM infiltration at days 32 to 35 after injection. (D) Quantification of BM infiltration in panel C. (E) Kaplan-Meier analyses of survival curves. Median survival (days): GFP+, 64; WT, 48; R209C, 40.5 (n = 15). Data are representative of 3 independent experiments with similar results. *P < .05, by log-rank test. (B,D) Data are expressed as the mean ± SD. *P < .05; **P < .01, by 2-tailed Student t test.
GNAO1 R209C mutation promotes ETV6-RUNX1+ leukemogenesis. (A) Representative images of the effects of ectopic expression of GNAO1 WT or R209C mutant on mouse spleen size at days 32 to 35 after injection. Reh cells expressing WT GNAO1, the R209C mutation, or a GFP control (GFP+) were injected into recipients through the tail vein. GFP−, negative GFP cell control. Images represent the results of 5 mice per group of 3 independent experiments. Scale bars, 1 cm. (B) Quantification of spleen weight in panel A. (C) Flow cytometry analysis of BM infiltration at days 32 to 35 after injection. (D) Quantification of BM infiltration in panel C. (E) Kaplan-Meier analyses of survival curves. Median survival (days): GFP+, 64; WT, 48; R209C, 40.5 (n = 15). Data are representative of 3 independent experiments with similar results. *P < .05, by log-rank test. (B,D) Data are expressed as the mean ± SD. *P < .05; **P < .01, by 2-tailed Student t test.
GNAO1 R209C mutation activates PI3K/Akt/mTOR signaling
Because Src-dependent activation of STAT-3 signaling is important for GNAO1 Q205L and RS43H mutation-induced NIH-3T3 cell transformation,10,19 we assessed whether the GNAO1 R209C mutation also regulates these signaling pathways. Compared with EV control, ectopic expression of WT GNAO1 or the R209C mutant significantly enhanced Akt and S6K phosphorylation (p-Akt and p-S6K) but not Erk1/2 or Src phosphorylation (Figure 5A). The R209C mutant induced much higher levels of p-Akt and p-S6K than that of WT GNAO1. This observation was further confirmed in the samples from the twins. Compared with expression in the PBMCs from twin B before the onset of leukemia, expression levels of GNAO1, p-Akt, and p-S6K were upregulated in the PBMCs from twin A at the onset of leukemia (Figure 5B). After leukemia remission, expression levels of GNAO1, p-Akt, and p-S6K were reduced in the PBMCs in twin A. Moreover, the PI3K inhibitor LY294002 significantly inhibited WT GNAO1 overexpression-induced p-Akt and p-S6K (Figure 5C) and R209C mutation overexpression-induced p-Akt, p-S6K, cell proliferation, and colony formation (Figure 5C; supplemental Figure 6A-B). The mTORC-1 inhibitor rapamycin reduced WT GNAO1 or R209C mutant overexpression-induced p-S6K but not p-Akt (Figure 5D). Overexpression of the GNAO1 R243C or Q307R mutation also increased GNAO1 activity and promoted cell proliferation, soft agar colony formation, and p-Akt, and p-S6K expression in Reh cells (supplemental Figure 7A-D). However, we found that ectopic expression of GNAO1 WT or the R209C mutant had no significant effects on expression of p-Akt and p-S6K in K562 cells with BCR/ABL fusion (supplemental Figure 5D). Taken together, our results demonstrate that GNAO1 R209C mutation-activated PI3K/Akt/mTOR signaling in leukemia depends on E/R fusion.
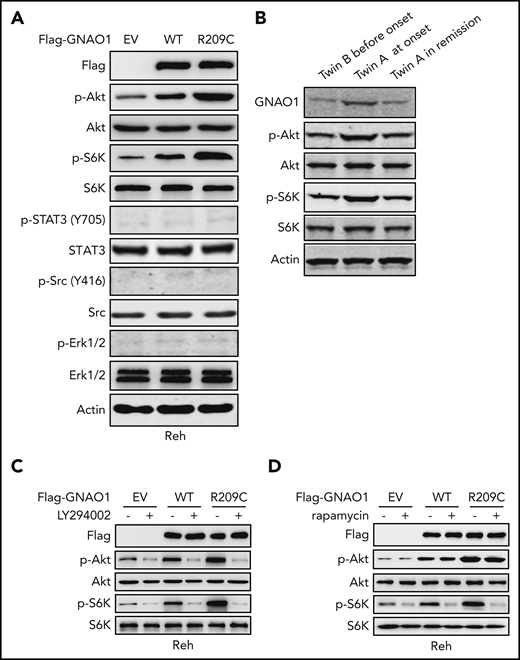
GNAO1 R209C mutation activates PI3K/Akt/mTOR signaling. (A) Western blot (WB) analysis of phosphorylation of Akt (p-Akt), STAT-3 (p-STAT3), Src (p-STAT3), and Erk1/2 (p-Erk1/2) in Reh cells with ectopic expression of Flag-tagged WT GNAO1, the R209C mutant, or an empty vector (EV) control. (B) WB of GNAO1, p-Akt, and p-Erk1/2 in the PBMC samples from twin B before onset, twin A at onset, and twin A at remission. (C) Effects of treatment with PI3K inhibitor LY294002 on p-Akt and p-S6K stimulation in Reh cells expressing WT GNAO1, the R209C mutant, or an EV control. Cells were treated with LY294002 (10 μM) for 1 hour. (D) Effects of treatment with the mTORC1 inhibitor rapamycin on p-Akt and p-S6K stimulation. Cells were treated with rapamycin (250 nM) for 1 hour. Data are representative of 3 independent experiments with similar results.
GNAO1 R209C mutation activates PI3K/Akt/mTOR signaling. (A) Western blot (WB) analysis of phosphorylation of Akt (p-Akt), STAT-3 (p-STAT3), Src (p-STAT3), and Erk1/2 (p-Erk1/2) in Reh cells with ectopic expression of Flag-tagged WT GNAO1, the R209C mutant, or an empty vector (EV) control. (B) WB of GNAO1, p-Akt, and p-Erk1/2 in the PBMC samples from twin B before onset, twin A at onset, and twin A at remission. (C) Effects of treatment with PI3K inhibitor LY294002 on p-Akt and p-S6K stimulation in Reh cells expressing WT GNAO1, the R209C mutant, or an EV control. Cells were treated with LY294002 (10 μM) for 1 hour. (D) Effects of treatment with the mTORC1 inhibitor rapamycin on p-Akt and p-S6K stimulation. Cells were treated with rapamycin (250 nM) for 1 hour. Data are representative of 3 independent experiments with similar results.
Reciprocal activation of E/R fusion and the GNAO1 R209C mutation induces leukemogenesis
The ETV6 gene encodes a transcriptional repressor,32,33 and the fusion of the ETV6 gene with RUNX1 gene generates a new oncogenic E/R transcription factor.3,4 Thus, we detected whether the expression of GNAO1 is affected by the E/R fusion gene. We collected 6 childhood ALL specimens, with or without E/R fusion. Using RT-PCR, we found that GNAO1 expression levels were significantly higher in E/R+ ALL specimens whereas less or no expression of GNAO1 was detected in E/R-negative samples (Figure 6A). Non-E/R fusion samples were also all identified as negative for E2A-PBX-1, m-BCR/ABL, MLL-AF4, inf-MLL-AF4, CML-related genes, M-BCR-ABL, AML-related genes, AML-ETO, L-PML-RARA, V-PML-RARA, S-PML-RARA, A-CBFB-MYH11, D-CBFB-MYH11, and E-CBFB-MYH11. To validate this observation, we collected another series of 8 ALL samples and 1 normal PBMC sample and confirmed that, compared with the normal PBMCs, GNAO1 protein expression was higher in most of the ALL specimens with E/R fusions but not in those without E/R fusions (Figure 6B). We then performed qRT-PCR analysis and found that, compared with those in normal PBMCs, GNAO1 expression levels were higher in E/R+ Reh and UoCB6 cells and in E/R− Nalm6 cells but not in THP-1 cells (Figure 6C). We further downloaded the GSE79869 data set and examined GNAO1 mRNA expression in normal PBMCs and ALL specimens with E/R fusion, hyperdiploid, or E2A-PBX1 fusion. The expression level of GNAO1 was significantly higher in ALL with E/R fusion than in normal PBMCs but not in other ALL samples (Figure 6D). These data suggest that GNAO1 expression is related to E/R fusion.
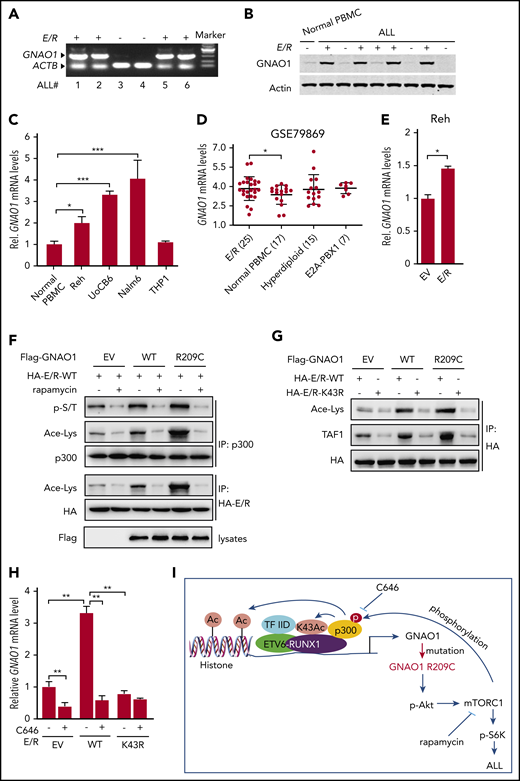
Reciprocal activation of E/R fusion and the GNAO1 R209C mutation induces leukemogenesis. (A) RT-PCR analysis of GNAO1 upregulation in ETV6-RUNX1 (E/R)+ clinical ALL specimens. ACTB was used as a control. (B) WB analysis of GNAO1 expression in normal PBMCs, E/R+, and E/R− childhood ALL specimens. (C) qRT-PCR analysis of GNAO1 mRNA expression in normal PBMCs, Reh, UoCB6, Nalm6, and THP1 leukemia cells. (D) The expression level of GNAO1 mRNA was significantly higher in E/R+ALL than in normal PBMCs. Expression data of GNAO1 mRNA were downloaded from the GSE79869 data set and analyzed. (E) qRT-PCR analysis of the effect of ectopic expression of the E/R fusion or an EV control on GNAO1 expression. (F) IP and WB analysis of the effects of rapamycin treatment on p300 acetylation (Ace-Lys), p300 serine/threonine phosphorylation (p-S/T), and E/R acetylation stimulated by ectopic expression of GNAO-1 WT or R209C mutation in Reh cells. (G) The K43R mutation of RUNX1 in E/R fusion inhibits E/R acetylation and association with TAF-1 stimulated by overexpression of GNAO1 WT or R209C mutant in Reh cells. (H) Treatment with the p300 inhibitor C464 reduces GNAO1 upregulation by E/R. (I) A hypothetical model showing how GNAO1 R209C mutation and E/R fusion cooperate to induce leukemia. Data are representative of 3 independent experiments with similar results. (C-E,H) Data are expressed as the mean ± SD. *P < .05; **P < .01; ***P < .001, by 2-tailed (C,E,H) or 1-tailed (D) Student t test.
Reciprocal activation of E/R fusion and the GNAO1 R209C mutation induces leukemogenesis. (A) RT-PCR analysis of GNAO1 upregulation in ETV6-RUNX1 (E/R)+ clinical ALL specimens. ACTB was used as a control. (B) WB analysis of GNAO1 expression in normal PBMCs, E/R+, and E/R− childhood ALL specimens. (C) qRT-PCR analysis of GNAO1 mRNA expression in normal PBMCs, Reh, UoCB6, Nalm6, and THP1 leukemia cells. (D) The expression level of GNAO1 mRNA was significantly higher in E/R+ALL than in normal PBMCs. Expression data of GNAO1 mRNA were downloaded from the GSE79869 data set and analyzed. (E) qRT-PCR analysis of the effect of ectopic expression of the E/R fusion or an EV control on GNAO1 expression. (F) IP and WB analysis of the effects of rapamycin treatment on p300 acetylation (Ace-Lys), p300 serine/threonine phosphorylation (p-S/T), and E/R acetylation stimulated by ectopic expression of GNAO-1 WT or R209C mutation in Reh cells. (G) The K43R mutation of RUNX1 in E/R fusion inhibits E/R acetylation and association with TAF-1 stimulated by overexpression of GNAO1 WT or R209C mutant in Reh cells. (H) Treatment with the p300 inhibitor C464 reduces GNAO1 upregulation by E/R. (I) A hypothetical model showing how GNAO1 R209C mutation and E/R fusion cooperate to induce leukemia. Data are representative of 3 independent experiments with similar results. (C-E,H) Data are expressed as the mean ± SD. *P < .05; **P < .01; ***P < .001, by 2-tailed (C,E,H) or 1-tailed (D) Student t test.
Next, to assess whether the E/R fusion regulates GNAO1 expression, we transfected the E/R into Reh cells and found that ectopic expression of the E/R upregulated GNAO1 mRNA expression (Figure 6E). We then performed an in silico analysis of a database of transcription factor–binding profiles (http://jaspar.genereg.net) and found that RUNX-1 may bind with the GNAO1 promoter at both sites: −1577 to −1567 and −586 to −576 (supplemental Figure 8A). To further investigate the role of E/R as a transcription factor of GNAO1, we performed ChIP-qPCR (ChIP-qPCR) on GNAO1, using antibodies directed against E/R to measure binding to the promoter of GNAO1 in Reh and UoCB6 cells and found that the E/R fusion bound to the GNAO1 promoter (supplemental Figure 8B). Moreover, overexpression of the E/R fusion activated GNAO1 promoter activity (supplemental Figure 8C). These data support that E/R fusion upregulates GNAO1 expression.
RUNX-1 has been demonstrated to be directly acetylated at the K43 residue by p300.34,35 Overexpression of p300 stimulates RUNX-1–dependent transcription and cell differentiation.35 Moreover, mTORC1 phosphorylates and activates p300.36 Thus, we hypothesized that E/R upregulates the expression of WT and mutated GNAO1, and the R209C mutation further enhances E/R transcriptional activity through mTORC1–activated p300 acetylation activity. To determine this, we performed IP and found that ectopic expression of GNAO1 R209C mutant markedly increased, not only the phosphorylation of Ser/Thr (p-S/T) and the acetylation (Ace-Lys) of p300, but also the Ace-Lys of E/R in Reh cells, compared with that in cells expressing the EV control or WT GNAO1 (Figure 6F). In contrast, rapamycin inhibited GNAO1 WT- or R209C mutant–stimulated p-S/T and Ace-Lys of p300 and Ace-Lys of E/R. To further assess whether p300-dependent acetylation promotes E/R transcriptional activity, we generated an E/R K43R mutation and overexpressed HA-tagged WT E/R and the K43R mutant in Reh cells. Ectopic expression of WT GNAO1 and the R209C mutant promoted E/R acetylation and association with TAF-1, a subunit of transcription factor protein complex TFIID37 (Figure 6G). RUNX-1 binding with TFIID is critical for its transcriptional activity.38 However, the E/R K43R mutation inhibited the E/R acetylation and association with TAF-1 stimulated by WT GNAO1 or the R209C mutant (Figure 6G). Moreover, ectopic expression of WT E/R but not the K43R mutant promoted GNAO1 mRNA expression, whereas treatment of the p300 inhibitor C646 markedly attenuated GNAO1 expression upregulated by WT E/R but not that by the K43R mutant (Figure 6H). Taken together, these data show that reciprocal activation of ETV-6-RUNX1 fusion and GNAO1 R209C mutation induces leukemogenesis by activating PI3K/Akt/mTOR signaling (Figure 6I).
Discussion
In this study, we functionally identified the GNAO1 R209C mutation as a novel second hit for E/R+ preleukemia. According to the 2-hit theory of leukemia, there should be a second hit of genetic damage that enables tumor cells to continuously maintain their heritability and malignant phenotype. In this research area, the study of monozygotic twins is one of the most direct and convenient strategies. By analyzing a pair of monozygotic twins discordant for childhood leukemia, we found that the frequency of E/R gene fusion was 12.5% and that the frequency of GNAO1-R209C mutation was only 0.35% in twin B’s sample during the healthy stage (2 months before onset), indicating that the prenatal first hit generated E/R+ preleukemia cells, and the second hit generated GNAO1 R209C–mutated leukemia cells in one fetus. After 2 months, the GNAO1 R209C mutation became the main clone at the onset of leukemia in twin B. Moreover, ectopic expression of the GNAO1 R209C mutant increased animal spleen size, weight, and BM infiltration in E/R+ Reh orthotopic xenograft-bearing mice and reduced the animal survival.
To the best of our knowledge, this is the first study to identify a GNAO1 mutant as an oncogene in human cancers. On the basis of previous studies, mutations of GNAO1 are often associated with epileptic encephalopathy and movement disorders.16,26,39 Although the GNAO1 R243H mutation was found in breast cancer20 and overexpression of the GNAO1 Q205L mutant promoted NIH-3T3 fibroblast neoplastic transformation in vitro,18 there has been no evidence to show that a GNAO1 mutant has been characterized as an oncogene in vivo. In this study, we identified the GNAO1 R209C mutant as a driver of childhood leukemia. Using whole-exome sequencing of a pair of monozygotic twins who were discordant for childhood leukemia, we first identified that GNAO1 R209C is a major point mutation that causes leukemogenesis in cooperation with E/R fusion. Similar to the GNAO1 Q205L mutation, the GNAO1 R209C mutation, as well as the R243C and Q307R mutations, were found to promote neoplastic transformation in cooperation with E/R. Finally, ectopic expression of the GNAO1 R209C mutant in leukemia cells with E/R fusion increased leukemia cell proliferation and colony formation in vitro and ALL tumorigenesis in vivo. Finding GNAO1 A166T and T329M mutations in leukemia with non-E/R fusion also indicates that these mutations may cooperate with other leukemia drivers or have different functions in leukemogenesis.
In this study, we demonstrate a new mechanism by which the GNAO1 R209C mutation activates PI3K/AKT/mTOR signaling in leukemia. GNAO1 mutations mediate GTPase activity and thereby inhibit or activate downstream signaling. For example, the constitutively active GNAO1 Q205L mutation promotes NIH-3T3 transformation by activating STAT-3 signaling.10 The constitutively active GNAO1 R243H mutation enhances Src and STAT-3 signaling in NIH-3T3 fibroblasts. In the present study, we showed that constitutive activation of the GNAO1 R209C mutation promotes PI3K/AKT/mTOR signaling. In leukemia cells, GNAO1 R209C mutant enhanced the activation of Akt and mTOR, whereas treatment with PI3K or mTOR inhibitor inhibited those targets, which is also consistent with the phenotypes of cell proliferation and tumorigenesis driven by ectopic expression of the GNAO-1 R209C mutant.
We also demonstrated that new reciprocal activation of E/R fusion and the GNAO1 R209C mutation induces leukemia. Whether the E/R fusion protein functions as a transcriptional repressor or an oncogenic transcriptional factor depends on the context. For example, the E/R fusion protein upregulates phosphatidylinositol 3-kinase VSP34, an important regulator of autophagy in leukemic human cells.40 In our study, we found that E/R fusion increased GNAO1 expression in leukemia cells in vitro, and the GNAO1 expression correlated positively with E/R fusion in clinical samples. Moreover, using genetic and pharmacological approaches, we demonstrated that GNAO1 R209C mutant promoted E/R transcriptional activity through acetylation of RUNX-1 in E/R at the K43 residue by mTORC1-phosphorylated p300, which is consistent with previous reports that RUNX-1 is acetylated at the K43 residue by p30034,35 and that mTORC1 phosphorylates and activates p300.36 Reciprocally, high activation of E/R further enhanced GNAO1 expression. This finding is in line with the clinical observation that the frequency of the GNAO1 R209C mutation increased from only 0.35% to ∼50% in 2 months in twin B. The molecular insight of this unique mechanism warrants further investigation.
In summary, although ETV6-RUNX1 fusion is recognized as a driver of B-cell ALL with additional molecular disruptions,2-4 the cooperative mechanisms are still unclear. Our mutational analysis, together with in vivo and in vitro functional assays, demonstrates that the GNAO1 R209C mutation functions as a second hit in facilitating both disease initiation and progression by activating PI3K/Akt/mTOR signaling before onset of leukemia with E/R fusion. Thus, GNAO1 mutation–mediated mTORC1–dependent activation of p300 may be an independent and distinct mechanism for potentiating leukemic transformation and progression in cooperation with another genetic abnormality. Nonetheless, the existence of GNAO1 mutations in a range of hematopoietic and nonhematopoietic malignancies suggests that the GNAO1 signaling pathway may be a common oncogenic mechanism in cancer, thereby offering a new opportunity for the development of effective cancer diagnostics and therapeutics.
All relevant data and methods supporting the findings of this study are available within the article and the supplementary Information or from the corresponding author on reasonable request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported, in part, by Shanghai Municipal Science and Technology Commission Grant 19JC1413500; National Natural Science Foundation of China Grants 81874078, 81972341, 81772663, 81670174, and 81670136; Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support 20161310; State Key Laboratory of Oncogenes and Related Genes in China grant zz-19-15; and Excellent Youth Scholar Initiation Grant 19XJ12003 (Shanghai Jiao Tong University).
Authorship
Contribution: H.F., B.L., and Y.L. designed and supervised the project; L.S., B.Y., Y.Y., J.L., Y.Z., L.D., T.W., X.W., S.W., and X.Y. performed experiments; L.S., B.Y., S.W., B.L., Y.L., and H.F. interpreted and/or reviewed the data; L.S., B.Y., Y.Y., J.T., Y.L., and H.F. wrote or edited the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Haizhong Feng, Renji-Med X Clinical Stem Cell Research Center, Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200127, China; e-mail: fenghaizhong@sjtu.edu.cn; Yanxin Li, Department of Hematology Oncology, Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai 200127, China; e-mail: liyanxin@scmc.com.cn; Benshang Li, Department of Hematology Oncology, Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai 200127, China; e-mail: leebenshang@hotmail.com; or Shengyue Wang, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200127, China; e-mail: wsy12115@rjh.com.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal