

Key Points
Cx62 is present in platelets and its inhibitor (62Gap27) attenuates hemichannel and gap junction–mediated intercellular communication.
62Gap27 inhibited platelet function, thrombosis, and hemostasis via upregulation of inhibitory PKA signaling in platelets.

Abstract
Connexins oligomerise to form hexameric hemichannels in the plasma membrane that can further dock together on adjacent cells to form gap junctions and facilitate intercellular trafficking of molecules. In this study, we report the expression and function of an orphan connexin, connexin-62 (Cx62), in human and mouse (Cx57, mouse homolog) platelets. A novel mimetic peptide (62Gap27) was developed to target the second extracellular loop of Cx62, and 3-dimensional structural models predicted its interference with gap junction and hemichannel function. The ability of 62Gap27 to regulate both gap junction and hemichannel-mediated intercellular communication was observed using fluorescence recovery after photobleaching analysis and flow cytometry. Cx62 inhibition by 62Gap27 suppressed a range of agonist-stimulated platelet functions and impaired thrombosis and hemostasis. This was associated with elevated protein kinase A–dependent signaling in a cyclic adenosine monophosphate–independent manner and was not observed in Cx57-deficient mouse platelets (in which the selectivity of 62Gap27 for this connexin was also confirmed). Notably, Cx62 hemichannels were observed to function independently of Cx37 and Cx40 hemichannels. Together, our data reveal a fundamental role for a hitherto uncharacterized connexin in regulating the function of circulating cells.
Introduction
Connexins constitute a family of channel-forming proteins that are distributed widely in different cell types.1-3 Connexins oligomerize in the endoplasmic reticulum to form 6-membered structures known as hemichannels that are transported to the plasma membrane. Hemichannels on adjacent cells dock together to form gap junctions or pore-like structures (∼2-3 nm) that facilitate contact-dependent intercellular trafficking of small molecules (up to 1 kDa) between adjacent cells, which enables coordinated cellular responses.1,4
Gap junctions and hemichannels have been studied in various cell types, where they mediate stable cellular interactions, and through mediation of intercellular signaling, they coordinate synchronized cell function within tissues.5 The cardiovascular functions of connexins are well-characterized, from cardiac myocyte contraction6-8 to control of vascular function.9-11 Notably, connexins such as connexin-37 (Cx37), Cx40, and Cx43 that are present in the vasculature have been reported to contribute to the development of atherosclerosis,12-14 a process in which circulating inflammatory cells are implicated.15-17 The reported roles of connexins in regulating the activities of immune cells, including monocytes, macrophages, T cells, and dendritic cells, in addition to platelets, are therefore particularly pertinent.18-21
Platelets are regulators of hemostasis and aggregate at sites of vascular damage to form thrombi that prevent excessive loss of blood.22,23 Increasing evidence indicates the importance of sustained signaling between platelets within a thrombus to ensure thrombus growth and stability and the importance of direct intercellular communication between adjacent platelets.24,25
We have reported the presence of Cx37 and Cx40 in platelets, and through the use of selective mimetic peptides and transgenic gene–deficient mice, we have demonstrated that both hemichannels and gap junctions are required for platelet activation and thrombus formation.26,27 In addition to Cx37 and Cx40, we observed notable levels of Cx62 messenger RNA transcripts in megakaryocytes and circulating cells such as B cells, T cells, and monocytes.26 Very little is known regarding the properties, function, and tissue distribution of this orphan connexin, which has previously been reported to be expressed only in the mouse (the mouse homolog is Cx57) in retina and muscle cells.28,29 Thus, we explored whether Cx62 has a role in platelets.
By using a newly designed inhibitory peptide (62Gap27) and Cx57-deficient mice, we reveal the importance of Cx62(57) for the regulation of intercellular signaling in platelets and within thrombi. Furthermore, we demonstrate that 62Gap27 inhibits platelet function by stimulating protein kinase A (PKA)–mediated inhibitory signaling that protects mice from thrombosis.
Methods
The preparation of washed platelets, immunoblotting, immunofluorescence, and platelet functional assays such as aggregation, dense granule secretion, fibrinogen binding, P-selectin exposure, calcium mobilization, clot retraction, platelet spreading, thrombus formation, and tail bleeding were performed as described previously.26,27,30-32 Detailed descriptions of reagents and these methods are provided in supplemental Methods (available on the Blood Web site).
Mice
Gja10em2(IMPC)Mbp mice were produced by insertion of an indel-causing frameshift mutation by the International Mouse Phenotyping Consortium (IMPC) at the University of California Davis; mice were also obtained in collaboration with the Mary Lyon Centre, Harwell, United Kingdom. Phenotyping of these mice in the IMPC pipeline revealed normal blood count parameters. C57BL/6 mice were purchased from Envigo (Huntingdon, United Kingdom).
Statistical analysis
Data were analyzed by using the Student t test, and if more than 2 means were present, significance was determined by 1-way analysis of variance (ANOVA), 2-way ANOVA (in vitro thrombus formation assay), nonparametric Mann-Whitney U test (in vivo thrombosis and amount of blood loss in tail-bleeding assay), and Fisher’s exact test (time to cessation of bleeding in tail bleeding assay). Data represent mean ± standard deviation, and P < .05 was considered to be statistically significant. Statistical analysis was performed using GraphPad Prism 7.0 software (San Diego, CA).
Results
Expression of Cx62 in platelets
The expression of Cx62 in human platelets and the megakaryocytic cell line MegO1 was confirmed by using immunoblot analysis, and the expression of Cx57 in mouse platelets was also observed (Figure 1A). HeLa cells are devoid of other connexin family members33 and were confirmed not to express detectable levels of Cx62. Immunofluorescence studies performed on resting permeabilized human platelets revealed that Cx62 (stained red) was present in a punctate arrangement inside platelets (stained green for GPIb) and was redistributed toward the periphery of the cells upon activation with the TxA2 mimetic peptide U46619 (used at a concentration at which platelet shape change is minimal) (Figure 1B-C).
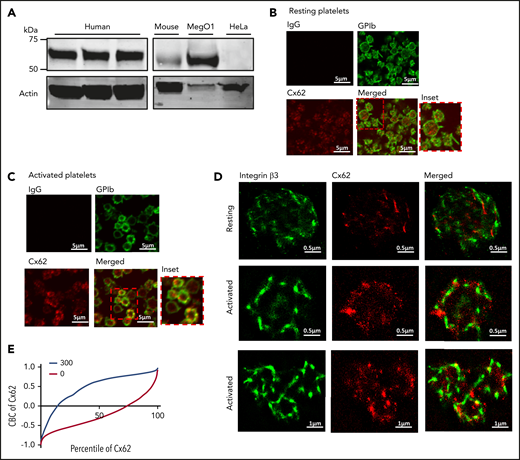
Expression and localization of Cx62 in platelets. (A) The presence of Cx62 was examined by immunoblot analysis of whole-cell lysates from human and mouse whole platelets, MegO1, and HeLa cells using a rabbit polyclonal anti-GJA10 antibody. Actin was used as a loading control. (B-C) The localization of Cx62 in resting and activated (with 5 μM U46619 in the presence of 3 μg/mL integrelin) permeabilized human platelets (0.2% Triton-X-100) was investigated using immunofluorescence microscopy. Cx62 (in red) and membrane GPIb receptors (in green) were stained using anti-GJA10 and anti-GPIb primary antibodies, respectively. Visualization was performed using Alexa-647– and Alexa-488–conjugated secondary antibodies, respectively. (D) The distribution of Cx62 was also studied using super-resolution stochastic optical reconstruction microscopy. Resting and activated permeabilized human platelets were stained using anti-GJA10 and anti-integrin β3 primary antibodies and visualized using Alexa-647– and Alexa-555–conjugated secondary antibodies, respectively. (E) CBC analysis was performed to determine the levels of Cx62 and β3 integrin colocalization in resting (0, red line) platelets and after stimulation with thrombin (1 U/mL) for 5 minutes (300, blue line). A CBC value of 0 represents a random distribution, and a positive value indicates closer distribution than expected at random. Data are representative of ≥3 separate experiments.
Expression and localization of Cx62 in platelets. (A) The presence of Cx62 was examined by immunoblot analysis of whole-cell lysates from human and mouse whole platelets, MegO1, and HeLa cells using a rabbit polyclonal anti-GJA10 antibody. Actin was used as a loading control. (B-C) The localization of Cx62 in resting and activated (with 5 μM U46619 in the presence of 3 μg/mL integrelin) permeabilized human platelets (0.2% Triton-X-100) was investigated using immunofluorescence microscopy. Cx62 (in red) and membrane GPIb receptors (in green) were stained using anti-GJA10 and anti-GPIb primary antibodies, respectively. Visualization was performed using Alexa-647– and Alexa-488–conjugated secondary antibodies, respectively. (D) The distribution of Cx62 was also studied using super-resolution stochastic optical reconstruction microscopy. Resting and activated permeabilized human platelets were stained using anti-GJA10 and anti-integrin β3 primary antibodies and visualized using Alexa-647– and Alexa-555–conjugated secondary antibodies, respectively. (E) CBC analysis was performed to determine the levels of Cx62 and β3 integrin colocalization in resting (0, red line) platelets and after stimulation with thrombin (1 U/mL) for 5 minutes (300, blue line). A CBC value of 0 represents a random distribution, and a positive value indicates closer distribution than expected at random. Data are representative of ≥3 separate experiments.
We also used super-resolution stochastic optical reconstruction microscopy (STORM) to determine the subcellular localization of Cx62. Compared with resting platelets, in activated platelets Cx62 (stained red) redistributed on (or near) the plasma membrane (stained green for integrin β3) and was arranged in clusters, thereby increasing proximity to integrin β3 in the plasma membrane (Figure 1D). Treatment with secondary antibody alone (in the absence of Cx62 primary antibody) was performed to exclude the possibility of nonspecific staining (supplemental Figure 1A). To further confirm the translocation of Cx62 to the plasma membrane upon platelet activation, colocalization of the integrin β3 subunit and Cx62 in resting and thrombin-stimulated platelets was analyzed using the coordinate-based colocalization (CBC) method.34 In CBC analysis, each molecule is assigned a value between −1 and 1. CBC values of zero indicate a homogeneous distribution of molecules, and positive values indicate increasing localization of the 2 sets of molecules. The shift in the CBC curve to predominantly positive values upon platelet stimulation, therefore indicates increased colocalization (Figure 1E). In nonstimulated platelets, ∼20% of the Cx62 population colocalized with integrin β3, which increased to ∼85% upon platelet activation (5 minutes) (Figure 1E).
To further explore the subcellular location of Cx62 in platelets, we performed a linear sucrose density gradient centrifugation on platelet homogenates after nitrogen cavitation. Cx62 was highly concentrated in the low-density fractions (1 and 2) with a distribution profile similar to that of calreticulin (a marker of the dense tubular system [DTS]) and β3 integrin but was absent from higher-density fractions (9 and 10), in which α-granule cargo such as TSP-1 was present (supplemental Figure 1B). These data are consistent with the presence of Cx62 inside and on the surface of platelets and further recruitment to the cell surface during platelet activation.
Cx62 structural prediction
To assist in the design and analysis of an inhibitory mimetic peptide that specifically targets Cx62(57), the monomeric and oligomeric structures of Cx62 were predicted. The predicted tertiary structure of Cx62 from the IntFOLD server35 reveals a protomer (monomer subunit) consisting of 4 transmembrane helices, 2 extracellular loops, a small bended N-terminal helix, and a long disordered cytoplasmic C-terminus loop (Figure 2A-B). The ModFOLD636 global 3-dimensional model quality score for the full-length protein was calculated as 0.43 (P < .01; <1/100 chance of being incorrect), which increased to 0.57 (P < .001; <1/1000 chance of being incorrect) when the long disordered C-terminal loop was excluded. The calculated local (or per residue) errors indicate that the ordered regions of the Cx62 structure were modeled with high confidence (Figure 2A). The tertiary structure model of Cx62 was subsequently used as a target for in silico docking with the designed mimetic peptide and for quaternary structure assembly of the docked hemichannel complex (Figure 2C-E).
![Design of the 62Gap27 mimetic peptide and its role in the regulation of intercellular communication. (A) Predicted 3-dimensional model of the Cx62 tertiary structure. The ribbon view of the structure is colored using the temperature coloring scheme in which blue indicates ordered regions with low predicted per-residue errors, and red indicates high per-residue errors and more flexibility. (B) Schematic representation of the designed 62Gap27 sequence on Cx62. In the topologic diagram of the Cx62 protomer, the predicted binding site is highlighted in orange. NT, NH2 terminus; CL, cytoplasmic loop; CT, COOH terminus; T, transmembrane; E, extracellular. (C) Surface representation of Cx62 hemichannels being targeted by 62Gap27 showing the pore cross-section and side views. (D) Inter-protomer interactions. The hemichannel formed by 6 protomers of Cx62 is shown in gray ribbon view, and the side chains in the zoomed views are shown as sticks with brown and yellow colors to differentiate between the residues of interacting protomer pairs. (E) Modeled intercellular interactions between docked hemichannels. In the left-hand panel, a Cx62 gap junction channel is shown. The region enclosed by dashed lines is sectioned perpendicular to the pore axis and is viewed from the pore axis (right-hand panel). The interactions between the 2 docked hemichannels (the first external loop [E1] and the second external loop [E2] regions) are depicted in the close-up images. In region E1, Gln58 forms symmetrical hydrogen bonds with the same residue from the opposite protomer, whereas Asn55 forms a hydrogen bond with Arg57 in the opposite protomer. In region E2, Asn196 and Asp199 form hydrogen bonds with the same residues on the opposite protomer. (F) The efflux of calcein from human platelets was measured using flow cytometric analysis. Calcein-loaded platelets incubated with 62Gap27 or scrambled peptide (100 μg/mL) were stimulated with thrombin (0.1 U/mL). Representative histograms of calcein fluorescence for unstimulated (green) and thrombin-stimulated platelets in the presence of scrambled peptide (blue) or 62Gap27 (100 µg/mL) (orange) (n = 4). (G) Calcein efflux after thrombin stimulation for varying time periods was measured by the rate of fluorescence reduction in platelets. Median fluorescence intensity for unstimulated and stimulated samples treated with scrambled peptide or 62Gap27 was analyzed (n = 4). (H) Calcein-loaded platelets were treated with scrambled peptide or 62Gap27 (100 µg/mL) for 5 minutes before their stimulation on fibrinogen and collagen-coated coverslips, and fluorescence recovery after photobleaching analysis was performed. Images represent fluorescence recovery (Pre-bleach, At-bleach, and Post-bleach) in samples treated with scrambled peptide or 62Gap27. (I) Quantified data show mean fluorescence recovery intensity of scrambled peptide and 62Gap27-treated samples normalized to the level of fluorescence at bleach point (shown in red circles; panel H) (n = 5). Data are mean ± standard error of the mean (SEM). ****P < .0001 (calculated by 2-way ANOVA).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/6/10.1182_blood.2019004575/3/m_bloodbld2019004575f2.png?Expires=1765883446&Signature=GCU-t~MEEHihSYqNrKl0Vpdkk6fAQCzFdVBHtuj8oJVFxxbUbKdrbjVCwDaOBqmhC-wmIdPLj8Y1M51nCFIyrBOVWV~2LzF3EWXmXbnbv8AT5NxnE9LrmZkLkgIyPtgUbBgcLTd~PQwuMjT~HkCLxDAxcQlcQq0oZ8ESFCtLi9NUDqYDPMm9~awoDzLo-HyysJQ7UikOPO2DxM3UR0s3LNCujpJH9uN-nmiv5zA168W-Iq9ljtqb6fWvz1JPOBV2z7YseQ-fgce-CRDb3TF012AozKdRoNJ5jhhGkdYeHgEmeSBs4jVfItgGPRVi~La6OBPQGuO54uvTDNeUdptQ0w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Design of the 62Gap27 mimetic peptide and its role in the regulation of intercellular communication. (A) Predicted 3-dimensional model of the Cx62 tertiary structure. The ribbon view of the structure is colored using the temperature coloring scheme in which blue indicates ordered regions with low predicted per-residue errors, and red indicates high per-residue errors and more flexibility. (B) Schematic representation of the designed 62Gap27 sequence on Cx62. In the topologic diagram of the Cx62 protomer, the predicted binding site is highlighted in orange. NT, NH2 terminus; CL, cytoplasmic loop; CT, COOH terminus; T, transmembrane; E, extracellular. (C) Surface representation of Cx62 hemichannels being targeted by 62Gap27 showing the pore cross-section and side views. (D) Inter-protomer interactions. The hemichannel formed by 6 protomers of Cx62 is shown in gray ribbon view, and the side chains in the zoomed views are shown as sticks with brown and yellow colors to differentiate between the residues of interacting protomer pairs. (E) Modeled intercellular interactions between docked hemichannels. In the left-hand panel, a Cx62 gap junction channel is shown. The region enclosed by dashed lines is sectioned perpendicular to the pore axis and is viewed from the pore axis (right-hand panel). The interactions between the 2 docked hemichannels (the first external loop [E1] and the second external loop [E2] regions) are depicted in the close-up images. In region E1, Gln58 forms symmetrical hydrogen bonds with the same residue from the opposite protomer, whereas Asn55 forms a hydrogen bond with Arg57 in the opposite protomer. In region E2, Asn196 and Asp199 form hydrogen bonds with the same residues on the opposite protomer. (F) The efflux of calcein from human platelets was measured using flow cytometric analysis. Calcein-loaded platelets incubated with 62Gap27 or scrambled peptide (100 μg/mL) were stimulated with thrombin (0.1 U/mL). Representative histograms of calcein fluorescence for unstimulated (green) and thrombin-stimulated platelets in the presence of scrambled peptide (blue) or 62Gap27 (100 µg/mL) (orange) (n = 4). (G) Calcein efflux after thrombin stimulation for varying time periods was measured by the rate of fluorescence reduction in platelets. Median fluorescence intensity for unstimulated and stimulated samples treated with scrambled peptide or 62Gap27 was analyzed (n = 4). (H) Calcein-loaded platelets were treated with scrambled peptide or 62Gap27 (100 µg/mL) for 5 minutes before their stimulation on fibrinogen and collagen-coated coverslips, and fluorescence recovery after photobleaching analysis was performed. Images represent fluorescence recovery (Pre-bleach, At-bleach, and Post-bleach) in samples treated with scrambled peptide or 62Gap27. (I) Quantified data show mean fluorescence recovery intensity of scrambled peptide and 62Gap27-treated samples normalized to the level of fluorescence at bleach point (shown in red circles; panel H) (n = 5). Data are mean ± standard error of the mean (SEM). ****P < .0001 (calculated by 2-way ANOVA).
Design of the 62Gap27 mimetic peptide and its role in the regulation of intercellular communication. (A) Predicted 3-dimensional model of the Cx62 tertiary structure. The ribbon view of the structure is colored using the temperature coloring scheme in which blue indicates ordered regions with low predicted per-residue errors, and red indicates high per-residue errors and more flexibility. (B) Schematic representation of the designed 62Gap27 sequence on Cx62. In the topologic diagram of the Cx62 protomer, the predicted binding site is highlighted in orange. NT, NH2 terminus; CL, cytoplasmic loop; CT, COOH terminus; T, transmembrane; E, extracellular. (C) Surface representation of Cx62 hemichannels being targeted by 62Gap27 showing the pore cross-section and side views. (D) Inter-protomer interactions. The hemichannel formed by 6 protomers of Cx62 is shown in gray ribbon view, and the side chains in the zoomed views are shown as sticks with brown and yellow colors to differentiate between the residues of interacting protomer pairs. (E) Modeled intercellular interactions between docked hemichannels. In the left-hand panel, a Cx62 gap junction channel is shown. The region enclosed by dashed lines is sectioned perpendicular to the pore axis and is viewed from the pore axis (right-hand panel). The interactions between the 2 docked hemichannels (the first external loop [E1] and the second external loop [E2] regions) are depicted in the close-up images. In region E1, Gln58 forms symmetrical hydrogen bonds with the same residue from the opposite protomer, whereas Asn55 forms a hydrogen bond with Arg57 in the opposite protomer. In region E2, Asn196 and Asp199 form hydrogen bonds with the same residues on the opposite protomer. (F) The efflux of calcein from human platelets was measured using flow cytometric analysis. Calcein-loaded platelets incubated with 62Gap27 or scrambled peptide (100 μg/mL) were stimulated with thrombin (0.1 U/mL). Representative histograms of calcein fluorescence for unstimulated (green) and thrombin-stimulated platelets in the presence of scrambled peptide (blue) or 62Gap27 (100 µg/mL) (orange) (n = 4). (G) Calcein efflux after thrombin stimulation for varying time periods was measured by the rate of fluorescence reduction in platelets. Median fluorescence intensity for unstimulated and stimulated samples treated with scrambled peptide or 62Gap27 was analyzed (n = 4). (H) Calcein-loaded platelets were treated with scrambled peptide or 62Gap27 (100 µg/mL) for 5 minutes before their stimulation on fibrinogen and collagen-coated coverslips, and fluorescence recovery after photobleaching analysis was performed. Images represent fluorescence recovery (Pre-bleach, At-bleach, and Post-bleach) in samples treated with scrambled peptide or 62Gap27. (I) Quantified data show mean fluorescence recovery intensity of scrambled peptide and 62Gap27-treated samples normalized to the level of fluorescence at bleach point (shown in red circles; panel H) (n = 5). Data are mean ± standard error of the mean (SEM). ****P < .0001 (calculated by 2-way ANOVA).
Design of the Cx62 mimetic peptide (62Gap27) and protein ligand docking studies
Because of the lack of an existing Cx62 selective inhibitor, we designed a mimetic peptide (62Gap27) that targets the second external loop of Cx62(57). To confirm the molecular interactions between Cx62 and 62Gap27, single ligand docking prediction was performed using SwissDock. Six of the clusters from SwissDock contained alternative ligand poses that were bound in approximately the same location at the end of the second external loop (Figure 2B) (supplemental Figure 1D).
Cx62 forms hemichannels and gap junctions in platelets
The exact mode of action by which different Gap27 peptides function is not clearly understood. It is believed that they induce a conformational change in the hemichannel that prevents them from docking to form a gap junction and thus modulate the permeability of the pore.21,26,37,38 To investigate this, we performed flow cytometry using calcein-loaded human platelets in which the efflux of calcein from the platelet cytosol was measured to determine the effect of 62Gap27 on Cx62 permeability (Figure 2F-G). Upon thrombin stimulation (0.1 U/mL), calcein-associated fluorescence decreased in scrambled peptide–treated cells by ∼50%, indicating a release of dye. The treatment of platelets with 62Gap27 (100 μg/mL) prevented this loss of fluorescence. Because flow cytometry–based analyses involve the gating of individual platelets, this indicates a role for Cx62 hemichannels in regulating platelet permeability. Given the strong reduction in the level of calcein efflux observed in 62Gap27-treated platelets, it is plausible that the peptide not only blocks Cx62 function but also inhibits the function of heteromers formed by Cx62 with other connexin isoforms present on platelets (eg, Cx37 and Cx40). At the same thrombin concentration, 62Gap27 did not reduce the extent of P-selectin exposure (a marker of α-granule secretion) on the platelet surface compared with treatment by scrambled peptides (supplemental Figure 1C). This suggests that the effects of 62Gap27 observed on the permeability of hemichannels were not a result of a reduction in the activation state of platelets under these experimental conditions.
To evaluate the ability of 62Gap27 to modulate gap junction–mediated intercellular communication, fluorescence recovery after photobleaching (FRAP) analysis was performed. Calcein-labeled platelets were incubated on coverslips coated with fibrinogen and collagen together (to ensure maximal platelet adhesion and spreading), and a defined region of cells was bleached by laser exposure. Fluorescence recovery in 62Gap27-treated platelet aggregates was halved compared with that in scrambled peptide–treated samples (17%) (Figure 2H-I). These findings demonstrate gap junction–mediated intercellular communication between platelets and the inhibitory effect of 62Gap27 on this.
The model of the Cx62 hemichannel complex (Figure 2C-D) revealed the 2 interacting hemichannels forming the putative structure of the Cx62 gap junction channel (Figure 2E). In the close-up view of the interface, the residues mediating the inter-hemichannel interactions are shown to be present in the first and second external loops (Figure 2E). Protomer-inhibitor (Cx62-62Gap27) interface residues were not found to coincide with the interface residues of the 12-mer (docked hemichannel). In addition, there are no SwissDock poses within the most common 62Gap27 inhibitor interaction location (Figure 2D) that share any interface residues with the with 6-mer (hemichannel) assembly (Figure 2D). Therefore, the inhibitor binding at this site is unlikely to disrupt either the assembly of the 6-mer or the hemichannel-hemichannel complex (12-mer) (Figure 2E).
The predicted 62Gap27 binding site was shown to coincide with the subsequent residues from which the inhibitor was designed (203-213, SRPTEKTIFML) (Figure 2B-C). Specifically, the inhibitor is likely to bind to both T209 and L213 (Figure 2B, bold circles). The additional interaction of the inhibitor with residues in the loop regions from 180 to 183 (GFQM) suggests a potential mechanism for the regulation of flow through the pore. The interaction may act to decrease the flexibility in the loop regions of the hemichannel pore, thereby regulating permeability (Figure 2C-D).
62Gap27 negatively regulates platelet aggregation and integrin activation
Light transmission aggregometry was used to investigate the effects of 62Gap27 on human washed platelets stimulated with a range of platelet activators that target different receptors. The concentrations of platelet agonists were optimized for each donor to attain 50% maximal aggregation (half-maximal effective concentration [EC50]) after 3 minutes of stimulation. Pretreatment of platelets with 62Gap27 (50 and 100 μg/mL) for 5 minutes caused a concentration-dependent inhibition to both CRP-XL (GPVI receptor–specific platelet agonist; EC50, 0.2-0.4 µg/mL) and thrombin-mediated (EC50, 0.05-0.08 U/mL) platelet aggregation (Figure 3A-B). The scrambled peptide (100 μg/mL) had no effect (supplemental Figure 2A). Inhibition of ∼45% (50 μg/mL 62Gap27) and 65% (100 μg/mL 62Gap27) was observed against CRP-XL and thrombin-stimulated aggregation, respectively. 62Gap27 also attenuated platelet aggregation stimulated by U46619 (EC50, 0.25-0.4 µM) (supplemental Figure 2B) and adenosine 5′-diphosphate (ADP) (EC50, 5-10 µM) (supplemental Figure 2C). These data suggest that the effects of 62Gap27, and therefore Cx62 functions, are common to a variety of platelet agonists.
![62Gap27 inhibits platelet activation and function specifically through Cx62. Washed human platelets (4 × 108 cells/mL) were treated with 62Gap27 or scrambled peptide (S; 100 μg/mL) for 5 minutes before their stimulation with (A) CRP-XL (EC50, 0.2-0.4 µg/mL) or (B) thrombin (EC50, 0.05-0.08 U/mL). Aggregation was measured using optical light transmission aggregometry for 180 seconds. Representative aggregation traces and quantified data shown (samples treated with scrambled peptide represent 100% aggregation). (C) Effects of 62Gap27 on CRP-XL (0.25 µg/mL) and thrombin (0.05 U/mL) mediated fibrinogen binding compared with the scrambled peptide (S; 100 μg/mL) was evaluated in platelets (in platelet-rich plasma [PRP]) using flow cytometry. (D) PRP from Cx57+/+ and Cx57−/− mice was treated with 62Gap27, 37,43Gap27, 40Gap27 (100 µg/mL), or scrambled peptide (S; 100 μg/mL) for 5 minutes. Fibrinogen binding levels were evaluated after stimulation with CRP-XL (1 µg/mL). (E) PRP from Cx57+/+ and Cx57−/− mice was stimulated with CRP-XL (1 µg/mL) and fibrinogen binding was measured. (F) P-selectin exposure was measured in 62Gap27 or scrambled peptide (S; 100 μg/mL) treated human platelets (in PRP) after stimulation with CRP-XL (0.25 μg/mL) or thrombin (0.05 U/mL). (G) Changes in ATP release were monitored for 5 minutes in washed platelets (4 × 108 cells/mL) incubated with 62Gap27 or scrambled peptide (S; 100 μg/mL) and stimulated with CRP-XL (0.5 µg/mL) or thrombin (0.05 U/mL). (H) The levels of TxB2 were measured by immunoassay in human washed human platelets (4 × 108 cells/mL) treated with scrambled peptide (S; 100 μg/mL) or 62Gap27 after stimulation with CRP-XL (0.5 µg/mL) or thrombin (0.05 U/mL). Data represent mean ± SEM (n ≥ 3). ns, not significant. *P < .05; **P < .01; ***P < .001 (calculated by 1-way ANOVA). †P < .05 (calculated by Student t test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/6/10.1182_blood.2019004575/3/m_bloodbld2019004575f3.png?Expires=1765883446&Signature=As2o7GGg444s2FZIu5Wc96E2KcVU0kS5QAl5KnCUXQ1WmAq6H1afIX0h51rkCnX4C3d60B8EEf2y8sB4uabh5uXnXUvkwnZBVkaC7p7ECRfwNRMxY3AX-LRXec3cj1Ejn~HxTV8rpl26mRY5tB6lWz-Oqu~dz-k7v38krG2zlIvi8lZjx6bY-LFFOyFr-NUx8zsoQBY2PUU-TPUXPPJGnVoHLxuACfPQF-ODyMD274grmDJIVdzVkgM4HPceAw92rt-Ia-~Q5GQ6PH3SuwC4IbeH6xhRcuzE4lLylxIS4Ugpq1EKEz2aVbGHlqA3DDxFZ8RsvtMmeyPoWUMRozdSWg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
62Gap27 inhibits platelet activation and function specifically through Cx62. Washed human platelets (4 × 108 cells/mL) were treated with 62Gap27 or scrambled peptide (S; 100 μg/mL) for 5 minutes before their stimulation with (A) CRP-XL (EC50, 0.2-0.4 µg/mL) or (B) thrombin (EC50, 0.05-0.08 U/mL). Aggregation was measured using optical light transmission aggregometry for 180 seconds. Representative aggregation traces and quantified data shown (samples treated with scrambled peptide represent 100% aggregation). (C) Effects of 62Gap27 on CRP-XL (0.25 µg/mL) and thrombin (0.05 U/mL) mediated fibrinogen binding compared with the scrambled peptide (S; 100 μg/mL) was evaluated in platelets (in platelet-rich plasma [PRP]) using flow cytometry. (D) PRP from Cx57+/+ and Cx57−/− mice was treated with 62Gap27, 37,43Gap27, 40Gap27 (100 µg/mL), or scrambled peptide (S; 100 μg/mL) for 5 minutes. Fibrinogen binding levels were evaluated after stimulation with CRP-XL (1 µg/mL). (E) PRP from Cx57+/+ and Cx57−/− mice was stimulated with CRP-XL (1 µg/mL) and fibrinogen binding was measured. (F) P-selectin exposure was measured in 62Gap27 or scrambled peptide (S; 100 μg/mL) treated human platelets (in PRP) after stimulation with CRP-XL (0.25 μg/mL) or thrombin (0.05 U/mL). (G) Changes in ATP release were monitored for 5 minutes in washed platelets (4 × 108 cells/mL) incubated with 62Gap27 or scrambled peptide (S; 100 μg/mL) and stimulated with CRP-XL (0.5 µg/mL) or thrombin (0.05 U/mL). (H) The levels of TxB2 were measured by immunoassay in human washed human platelets (4 × 108 cells/mL) treated with scrambled peptide (S; 100 μg/mL) or 62Gap27 after stimulation with CRP-XL (0.5 µg/mL) or thrombin (0.05 U/mL). Data represent mean ± SEM (n ≥ 3). ns, not significant. *P < .05; **P < .01; ***P < .001 (calculated by 1-way ANOVA). †P < .05 (calculated by Student t test).
62Gap27 inhibits platelet activation and function specifically through Cx62. Washed human platelets (4 × 108 cells/mL) were treated with 62Gap27 or scrambled peptide (S; 100 μg/mL) for 5 minutes before their stimulation with (A) CRP-XL (EC50, 0.2-0.4 µg/mL) or (B) thrombin (EC50, 0.05-0.08 U/mL). Aggregation was measured using optical light transmission aggregometry for 180 seconds. Representative aggregation traces and quantified data shown (samples treated with scrambled peptide represent 100% aggregation). (C) Effects of 62Gap27 on CRP-XL (0.25 µg/mL) and thrombin (0.05 U/mL) mediated fibrinogen binding compared with the scrambled peptide (S; 100 μg/mL) was evaluated in platelets (in platelet-rich plasma [PRP]) using flow cytometry. (D) PRP from Cx57+/+ and Cx57−/− mice was treated with 62Gap27, 37,43Gap27, 40Gap27 (100 µg/mL), or scrambled peptide (S; 100 μg/mL) for 5 minutes. Fibrinogen binding levels were evaluated after stimulation with CRP-XL (1 µg/mL). (E) PRP from Cx57+/+ and Cx57−/− mice was stimulated with CRP-XL (1 µg/mL) and fibrinogen binding was measured. (F) P-selectin exposure was measured in 62Gap27 or scrambled peptide (S; 100 μg/mL) treated human platelets (in PRP) after stimulation with CRP-XL (0.25 μg/mL) or thrombin (0.05 U/mL). (G) Changes in ATP release were monitored for 5 minutes in washed platelets (4 × 108 cells/mL) incubated with 62Gap27 or scrambled peptide (S; 100 μg/mL) and stimulated with CRP-XL (0.5 µg/mL) or thrombin (0.05 U/mL). (H) The levels of TxB2 were measured by immunoassay in human washed human platelets (4 × 108 cells/mL) treated with scrambled peptide (S; 100 μg/mL) or 62Gap27 after stimulation with CRP-XL (0.5 µg/mL) or thrombin (0.05 U/mL). Data represent mean ± SEM (n ≥ 3). ns, not significant. *P < .05; **P < .01; ***P < .001 (calculated by 1-way ANOVA). †P < .05 (calculated by Student t test).
Flow cytometry was used to measure the level of fibrinogen binding to activated integrin αIIbβ3. Consistent with reduced aggregation, CRP-XL or thrombin-stimulated fibrinogen binding was reduced by 55% and 65%, respectively, after treatment with 62Gap27 (100 μg/mL) (Figure 3C). The scrambled peptide had no effect (supplemental Figure 2D). Fibrinogen binding was measured on gated single platelets, which provides additional evidence that Cx62 hemichannels participate in the initiation of platelet activation.
The actions of 62Gap27 are mediated selectively via Cx62
To confirm the selectivity of 62Gap27 mimetic peptide toward Cx62(57), its effects were examined in Cx57−/− platelets. The deletion of Cx57 did not alter the expression of other platelet connexins such as Cx37 and Cx40 (supplemental Figure 3A-B). Similarly, the deletion of Cx37 and Cx40 did not affect the expression of Cx57 (supplemental Figure 3C-D). The expression of GPVI, integrin α2β1, integrin αIIbβ3, and GPIb on the surface of Cx57+/+ and Cx57−/− platelets was not significantly different (supplemental Figure 3E-H).
Compared with scrambled peptide, treatment with 62Gap27 (100 µg/mL) inhibited CRP-XL–mediated fibrinogen binding in Cx57+/+ platelets (in platelet-rich plasma) but had no effect on Cx57−/− platelets (Figure 3D), confirming the specificity of 62Gap27 for Cx57, which in turn signifies its selectivity for Cx62 in humans. In addition, 37,43Gap27 and 40Gap27 treatment (mimetic peptides for Cx37 and Cx40, respectively) significantly inhibited fibrinogen binding in both Cx57+/+ and Cx57−/− platelets (Figure 3D). Consistent with this, 62Gap27 also inhibited CRP-XL–stimulated fibrinogen binding in Cx37−/− and Cx40−/− platelets to an extent similar to that in littermate Cx37+/+ and Cx40+/+ platelets (supplemental Figure 4A-B). This suggests that Cx37, Cx40, and Cx57 hemichannels can function independently of each other in platelets, and the deletion of 1 connexin does not affect the functions of other platelet connexins. Furthermore, a significant reduction in fibrinogen binding was observed in CRP-XL–stimulated Cx57−/− platelets compared with Cx57+/+ platelets. This indicates a fundamental role of Cx57 in regulating platelet activation (Figure 3E).
62Gap27 inhibits platelet secretion
To assess the effects of 62Gap27 on platelet secretion, P-selectin surface exposure and adenosine triphosphate (ATP) release (a marker of dense granule secretion) were evaluated by using flow cytometry and luciferin-luciferase luminescence assay, respectively. Incubation of platelets (in platelet-rich plasma) with 62Gap27 attenuated (compared with scrambled peptide) P-selectin exposure, reaching 60% inhibition (at 100 μg/mL 62Gap27) upon stimulation with CRP-XL or thrombin (Figure 3F). Scrambled peptide (100 μg/mL) had no effect (supplemental Figure 4C). CRP-XL or thrombin-stimulated ATP release was also attenuated by ∼65% and 50%, respectively, upon treatment with 62Gap27 (100 μg/mL) compared with the scrambled peptide (Figure 3G).
Integrin-mediated platelet adhesion and signaling is regulated by Cx62
Binding of fibrinogen to integrin αIIbβ3 initiates integrin clustering and outside-in signaling that reinforces platelet activation and ensures the stability of the thrombus.40 Platelet spreading and clot retraction are direct outcomes of outside-in signaling.41 The effects of 62Gap27 on platelet adhesion and spreading on fibrinogen coated-coverslips were investigated. Compared with the scrambled peptide–treated controls, 62Gap27 (50 and 100 μg/mL) significantly reduced platelet adhesion to fibrinogen (Figure 4A). The proportion of adhered platelets reaching lamellipodia stage was also downregulated by 62Gap27 (Figure 4A). In the absence of agonist prestimulation, the ability of 62Gap27 to attenuate platelet adhesion to fibrinogen-coated coverslips suggests underlying levels of platelet signaling that are associated with Cx62 function and can modulate integrin affinity and fibrinogen binding. Consistent with this, fibrin clot retraction was also inhibited, indicating the role of Cx62 in the formation and consolidation of thrombi (Figure 4B).

62Gap27 inhibits integrin αIIbβ3-mediated signaling, thrombosis, and hemostasis. (A) Human washed platelets (2 × 107 cells/mL) incubated for 5 minutes with 62Gap27 (50 and 100 μg/mL) or scrambled peptide (S; 100 μg/mL) were exposed to fibrinogen-coated (100 μg/mL) coverslips. Representative images of spreading and adhesion of platelets after 45 minutes and cumulative data for platelets adhered to fibrinogen in each sample are shown. Spreading platelets were categorized into 3 groups (adhered but not spread; filopodia, platelets in the process of extending filopodia; and lamellipodia, fully spread). (B) To measure clot retraction, human PRP was incubated with 62Gap27 (50 and 100 μg/mL) or scrambled peptide (S; 100 μg/mL) for 5 minutes before clot formation was initiated by the addition of thrombin (1 U/mL). The extent of clot retraction was determined by comparing clot weight after 60 minutes. (C) 3,3′-Dihexyloxacarbocyanine iodide (DiOC6)-loaded human whole blood was treated with scrambled peptide or 62Gap27 (100 μg/mL) for 5 minutes before perfusion through collagen-coated (100 μg/mL) Vena8Biochips at an arterial shear rate of 500 s−1 (20 dyne/cm2). Representative images display thrombus formation at the end of the assay (10 minutes), and quantified data represent mean thrombus fluorescence intensity. (D) In vivo thrombosis was assayed by intravital microscopy after the laser-induced injury. 62Gap27 or scrambled peptide (100 μg/mL) was administered intravenously to mice, and platelets were fluorescently labeled with Alexa-647–conjugated anti-GPIb antibody. After laser injury, platelet accumulation and thrombus formation were assessed. Representative images at different time points are shown, and data are expressed as median fluorescence intensity. (E) The mean of maximum fluorescence intensity was calculated from the maximum fluorescence intensity of each thrombi. A total of 21 thrombi were analyzed from 5 mice treated for each condition. (F) Tail bleeding as determined by time to cessation of bleeding in mice treated with scrambled peptide (S) or 62Gap27 (100 μg/mL) for 5 minutes (mice treated with scrambled peptide, n = 9; samples treated with 62Gap27, n = 10). (G) The amount of blood loss was evaluated after the cessation of tail bleeding in mice treated with scrambled peptide or 62Gap27 (100 μg/mL) for 5 minutes. Data represent mean ± SEM (n ≥ 3). P values were calculated by 1-way ANOVA (spreading assay), 2-way ANOVA (in vitro thrombus formation assay), nonparametric Mann-Whitney U test (in vivo thrombosis and blood loss in tail-bleeding assay), and Fisher’s exact test (time to cessation of bleeding in tail bleeding assay). *P < .05; **P < .01; ***P < .001; ****P < .0001.
62Gap27 inhibits integrin αIIbβ3-mediated signaling, thrombosis, and hemostasis. (A) Human washed platelets (2 × 107 cells/mL) incubated for 5 minutes with 62Gap27 (50 and 100 μg/mL) or scrambled peptide (S; 100 μg/mL) were exposed to fibrinogen-coated (100 μg/mL) coverslips. Representative images of spreading and adhesion of platelets after 45 minutes and cumulative data for platelets adhered to fibrinogen in each sample are shown. Spreading platelets were categorized into 3 groups (adhered but not spread; filopodia, platelets in the process of extending filopodia; and lamellipodia, fully spread). (B) To measure clot retraction, human PRP was incubated with 62Gap27 (50 and 100 μg/mL) or scrambled peptide (S; 100 μg/mL) for 5 minutes before clot formation was initiated by the addition of thrombin (1 U/mL). The extent of clot retraction was determined by comparing clot weight after 60 minutes. (C) 3,3′-Dihexyloxacarbocyanine iodide (DiOC6)-loaded human whole blood was treated with scrambled peptide or 62Gap27 (100 μg/mL) for 5 minutes before perfusion through collagen-coated (100 μg/mL) Vena8Biochips at an arterial shear rate of 500 s−1 (20 dyne/cm2). Representative images display thrombus formation at the end of the assay (10 minutes), and quantified data represent mean thrombus fluorescence intensity. (D) In vivo thrombosis was assayed by intravital microscopy after the laser-induced injury. 62Gap27 or scrambled peptide (100 μg/mL) was administered intravenously to mice, and platelets were fluorescently labeled with Alexa-647–conjugated anti-GPIb antibody. After laser injury, platelet accumulation and thrombus formation were assessed. Representative images at different time points are shown, and data are expressed as median fluorescence intensity. (E) The mean of maximum fluorescence intensity was calculated from the maximum fluorescence intensity of each thrombi. A total of 21 thrombi were analyzed from 5 mice treated for each condition. (F) Tail bleeding as determined by time to cessation of bleeding in mice treated with scrambled peptide (S) or 62Gap27 (100 μg/mL) for 5 minutes (mice treated with scrambled peptide, n = 9; samples treated with 62Gap27, n = 10). (G) The amount of blood loss was evaluated after the cessation of tail bleeding in mice treated with scrambled peptide or 62Gap27 (100 μg/mL) for 5 minutes. Data represent mean ± SEM (n ≥ 3). P values were calculated by 1-way ANOVA (spreading assay), 2-way ANOVA (in vitro thrombus formation assay), nonparametric Mann-Whitney U test (in vivo thrombosis and blood loss in tail-bleeding assay), and Fisher’s exact test (time to cessation of bleeding in tail bleeding assay). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Cx62(57) modulates thrombosis and hemostasis
To elucidate the function of Cx62 in platelets under shear in whole blood, we determined the effects of 62Gap27 on thrombus formation in vitro using fluorescence microscopy. 3,3′-Dihexyloxacarbocyanine iodide (DiOC6)–labeled whole human blood treated with scrambled peptide or 62Gap27 (100 μg/mL), was perfused through collagen-coated microfluidic flow channels for 10 minutes at a shear rate of 500 s−1 (20 dyn/cm2). The mean fluorescence intensity of thrombi in 62Gap27-treated whole blood was reduced by 70% compared with scrambled peptide (Figure 4C). Furthermore, the extent of thrombus surface coverage was also attenuated in 62Gap27-treated samples, consistent with impaired adhesion of platelets (supplemental Figure 4D).
The acute effects of 62Gap27 (100 μg/mL) on thrombosis was investigated in mice after laser-induced injury of cremaster muscle arterioles and was observed using intravital microscopy. Large and stable thrombi were formed in mice treated with scrambled peptide, whereas 62Gap27 treatment resulted in the development of substantially smaller thrombi that were unstable and embolized, resulting in a threefold reduction in the mean of maximum fluorescence intensity (Figure 4D-E).
The contribution of Cx57(62) to hemostasis was assessed by measuring tail-bleeding time. Although bleeding stopped in all 9 mice treated with scrambled peptide (mean bleeding time, 459 ± 81 seconds), the time to cessation of bleeding was dramatically increased in mice treated with 62Gap27; 7 of 10 mice bled for more than 20 minutes (Figure 4F). In alignment with this, the amount of blood loss in mice treated with 62Gap27 was higher than that in mice treated with the scrambled peptide, indicating impaired hemostasis (Figure 4G).
62Gap27 negatively regulates GPVI and thrombin-mediated signaling in platelets
Given the effects of 62Gap27 on CRP-XL–mediated platelet responses and thrombus formation in vitro and in vivo, we investigated the effects of 62Gap27 on signal transduction stimulated by the GPVI receptor. Pretreatment of platelets (nonaggregation conditions) with 62Gap27 (50 and 100 µg/mL) for 5 minutes reduced CRP-XL–stimulated total protein tyrosine phosphorylation by approximately 25% and 30%, respectively, compared with scrambled peptide (Figure 5A). Consistent with this, 100 µg/mL of 62Gap27 inhibited the tyrosine phosphorylation of Syk (Tyr525/526) by ∼30% (Figure 5B), which indicated that 62Gap27 inhibits the early stages of the GPVI signaling. Activated Syk results in phosphorylation of the transmembrane adapter protein LAT followed by PLCγ2 phosphorylation.41 Tyrosine phosphorylation of LAT (Tyr200) and PLCγ2 (Tyr1217) were observed to be diminished by 40% and 25%, respectively, after treatment with 62Gap27 (100 µg/mL) compared with the scrambled peptide (Figure 5C-D).
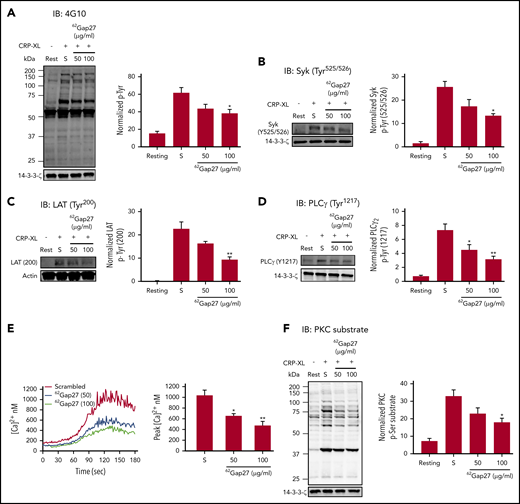
62Gap27 inhibits GPVI signaling in human platelets.62Gap27 (50 and 100 μg/mL) or scrambled peptide (S; 100 μg/mL) pretreated human washed platelets (4 × 108 cells/mL) were stimulated for 90 seconds with CRP-XL (1 μg/mL) under nonaggregation conditions in the presence of indomethacin (20 µM), cangrelor (1 µM), MRS2179 (100 µM), and EGTA (1 mM). Samples were lysed in the Laemmli sample buffer, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene fluoride (PVDF) membranes, and tested for (A) total tyrosine phosphorylation, (B) Syk phosphorylation (Tyr525/526), (C) LAT phosphorylation (Tyr200), (D) PLCγ phosphorylation (Tyr1217), and (E) Fura-2-acetoxymethyl ester (Fura-2AM)–loaded washed platelets (4 × 108 cells/mL) were treated with 62Gap27 or scrambled peptide (S; 100 μg/mL) for 5 minutes before stimulation with CRP-XL (0.25 μg/mL). Spectrofluorimetry was used to measure the release of calcium from intracellular stores. (F) PKC substrate phosphorylation. Representative blots for the phosphorylation levels are shown. The bar graph represents mean normalized phosphorylation values relative to actin or 14-3-3-ζ levels. Representative traces of calcium mobilization over a period of 5 minutes and quantified data (peak calcium levels) are shown. Results are mean ± SEM (n ≥ 3). *P < .05; **P < .01 (calculated by 1-way ANOVA).
62Gap27 inhibits GPVI signaling in human platelets.62Gap27 (50 and 100 μg/mL) or scrambled peptide (S; 100 μg/mL) pretreated human washed platelets (4 × 108 cells/mL) were stimulated for 90 seconds with CRP-XL (1 μg/mL) under nonaggregation conditions in the presence of indomethacin (20 µM), cangrelor (1 µM), MRS2179 (100 µM), and EGTA (1 mM). Samples were lysed in the Laemmli sample buffer, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene fluoride (PVDF) membranes, and tested for (A) total tyrosine phosphorylation, (B) Syk phosphorylation (Tyr525/526), (C) LAT phosphorylation (Tyr200), (D) PLCγ phosphorylation (Tyr1217), and (E) Fura-2-acetoxymethyl ester (Fura-2AM)–loaded washed platelets (4 × 108 cells/mL) were treated with 62Gap27 or scrambled peptide (S; 100 μg/mL) for 5 minutes before stimulation with CRP-XL (0.25 μg/mL). Spectrofluorimetry was used to measure the release of calcium from intracellular stores. (F) PKC substrate phosphorylation. Representative blots for the phosphorylation levels are shown. The bar graph represents mean normalized phosphorylation values relative to actin or 14-3-3-ζ levels. Representative traces of calcium mobilization over a period of 5 minutes and quantified data (peak calcium levels) are shown. Results are mean ± SEM (n ≥ 3). *P < .05; **P < .01 (calculated by 1-way ANOVA).
Downstream of PLCγ2, 62Gap27 inhibited CRP-XL–stimulated calcium mobilization by 45% compared with scrambled peptide (Figure 5E). Treatment with saturating concentrations of EGTA (2 mM) to prevent Ca2+ influx in the presence of scrambled peptide reduced a CRP-XL–mediated rise in cytosolic calcium concentration by ∼50% compared with scrambled peptide alone. The inhibitory effects of 62Gap27 (100 μg/mL) were found to be additive to the reduction caused by EGTA-scrambled peptide after stimulation with CRP-XL (supplemental Figure 5A), suggesting that Cx62 can modulate the release of calcium from intracellular stores. Consistent with this, CRP-XL–evoked PKC substrate phosphorylation was also found to be attenuated by 45% (Figure 5F) after incubation with 62Gap27 (100 μg/mL). Furthermore, 62Gap27 also reduced ERK1/2 phosphorylation in CRP-XL–stimulated platelets, which is consistent with the downregulation of CRP-XL–evoked TxA2 (TxB2) release (supplemental Figure 5F). Similar inhibition was observed after treatment with 37,43Gap27 (supplemental Figure 5F).
Compared with scrambled peptide, 62Gap27 also inhibited thrombin-evoked total protein tyrosine phosphorylation, calcium mobilization, the release of calcium from intracellular stores, PKC substrate phosphorylation, and ERK1/2 phosphorylation (supplemental Figure 5B-F).
To confirm that signaling events after GPVI activation were affected by Cx57 in mice, GPVI signaling events were investigated in Cx57+/+ and Cx57−/− platelets. Cx57-deficient platelets showed reduced CRP-XL-evoked total tyrosine phosphorylation and phosphorylation of Syk (Tyr525/526), LAT (Tyr200), PLCγ2 (Tyr1217), and PKC substrates compared with Cx57+/+ mouse platelets (Figure 6A-E), suggesting the importance of Cx57 in GPVI signaling.
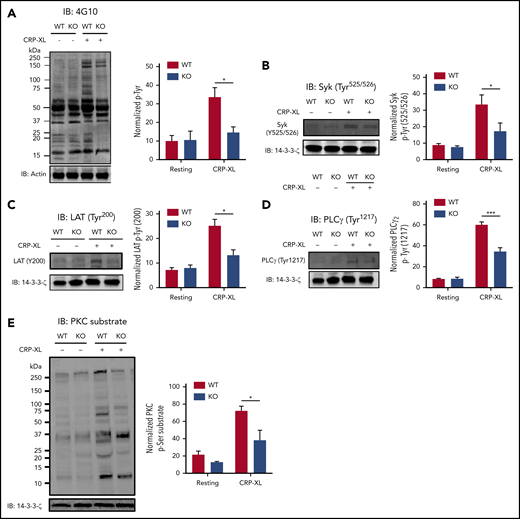
Deletion of Cx57 impairs GPVI signaling in platelets.62Gap27 (50 and 100 μg/mL) or scrambled peptide (S; 100 μg/mL) pretreated Cx57+/+ wild-type (WT) and Cx57−/− knockout (KO) washed platelets (4 × 108 cells/mL) were stimulated for 90 seconds with CRP-XL (1 μg/mL) under nonaggregation conditions in the presence of indomethacin (20 µM), cangrelor (1 µM), MRS2179 (100 µM), and EGTA (1 mM). Samples were lysed in the Laemmli sample buffer, separated by SDS-PAGE, transferred to PVDF membranes, and tested for (A) total tyrosine phosphorylation, (B) Syk phosphorylation (Tyr525/526), (C) LAT phosphorylation (Tyr200), (D) PLCγ phosphorylation (Tyr1217), and (E) PKC substrate phosphorylation. Representative blots for the phosphorylation levels are shown. The bar graph represents mean normalized phosphorylation values relative to actin or 14-3-3-ζ levels. Results are mean ± SEM (n ≥ 3). *P < .05; ***P < .001 (calculated by 1-way ANOVA).
Deletion of Cx57 impairs GPVI signaling in platelets.62Gap27 (50 and 100 μg/mL) or scrambled peptide (S; 100 μg/mL) pretreated Cx57+/+ wild-type (WT) and Cx57−/− knockout (KO) washed platelets (4 × 108 cells/mL) were stimulated for 90 seconds with CRP-XL (1 μg/mL) under nonaggregation conditions in the presence of indomethacin (20 µM), cangrelor (1 µM), MRS2179 (100 µM), and EGTA (1 mM). Samples were lysed in the Laemmli sample buffer, separated by SDS-PAGE, transferred to PVDF membranes, and tested for (A) total tyrosine phosphorylation, (B) Syk phosphorylation (Tyr525/526), (C) LAT phosphorylation (Tyr200), (D) PLCγ phosphorylation (Tyr1217), and (E) PKC substrate phosphorylation. Representative blots for the phosphorylation levels are shown. The bar graph represents mean normalized phosphorylation values relative to actin or 14-3-3-ζ levels. Results are mean ± SEM (n ≥ 3). *P < .05; ***P < .001 (calculated by 1-way ANOVA).
62Gap27 activates PKA independently of cyclic nucleotide signaling
The effects of 62Gap27 on GPVI-specific signaling events were surprising, given the ability of the peptide to inhibit platelet responses to several agonists including thrombin, which suggest a mechanism that would be common to each. Platelets are maintained in a quiescent state by prostaglandin I2 (PGI2) and nitric oxide (NO) released by endothelial cells.42,43 PGI2 binds to an IP receptor and stimulates the production of cyclic adenosine monophosphate (cAMP), whereas NO stimulates the production of cyclic guanosine monophosphate (cGMP), which activates PKA and PKG, respectively, and inhibits platelet activation through phosphorylation of multiple substrates.44,45
We therefore explored the effect of 62Gap27 on PKA- and PKG-dependent signaling in platelets by measuring the extent of vasodilator-stimulated phosphoprotein (VASP) phosphorylation at Ser157 and Ser239, respectively, (PKA- and PKG-selective phosphorylation sites). The treatment of resting platelets with 62Gap27 upregulated the phosphorylation of VASP Ser157 compared with scrambled peptide (Figure 7A), whereas no effect on VASP Ser239 was observed (supplemental Figure 6A). VASP Ser157 phosphorylation was also elevated in CRP-XL–stimulated platelets that were treated with 62Gap27 compared with control treated with scrambled peptide (Figure 7B). In addition, we observed that resting platelets, when treated with 37,43Gap27, also upregulate VASP Ser157 phosphorylation compared with scrambled peptide (supplemental Figure 6B). This suggests that activation of PKA represents a general mechanism by which Gap27 peptides inhibit platelet functions.
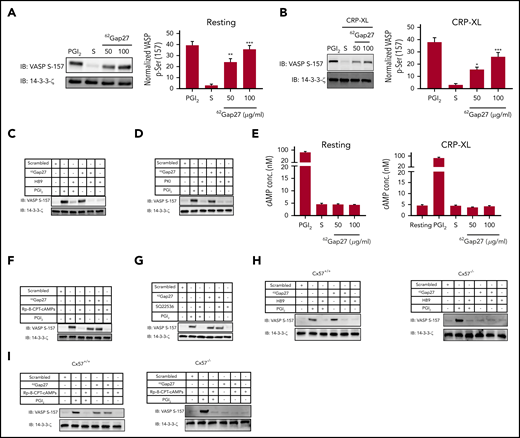
62Gap27 modulates PKA activity independently of cAMP. (A) Resting and (B) CRP-XL stimulated (90 seconds) washed human platelets (4 × 108 cells/mL) treated with scrambled peptide (S; 100 µg/mL) or 62Gap27 (50 and 100 μg/mL) for 5 minutes were tested for VASP S157 phosphorylation (a marker of PKA activity). VASP S157 phosphorylation was also evaluated in washed platelets treated with 62Gap27 (100 μg/mL) for 5 minutes in the presence of (C) H89 (10 µM), (D) PKI (10 µM), (F) Rp-8-CPT-cAMPS (200 µM), and (G) SQ 22536 (100 µM). Platelets treated with PGI2 (1 μg/mL) for the stimulation of PKA-mediated phosphorylation were used as positive controls. Sample lysis was carried out using the Laemmli sample buffer before separation by SDS-PAGE. Samples were then transferred to PVDF membranes. 14-3-3-ζ was used as a loading control. (E) Levels of cAMP were measured in resting and CRP-XL (1 μg/mL) stimulated washed human platelets (4 × 108 cells/mL) that had been preincubated with the scrambled peptide (S; 100 µg/mL) or 62Gap27 (50 or 100 µg/mL) for 5 minutes. Platelets treated with PGI2 (1 μg/mL) were used as a positive control. (H-I) Resting washed platelets (4 × 108 cells/mL) from Cx57+/+ and Cx57−/− mice were treated with scrambled peptide (S) or 62Gap27 (100 μg/mL) for 5 minutes in the presence of H89 (10 µM) or Rp-8-CPT-cAMPS (200 µM) and investigated for VASP-S157 phosphorylation. Platelets treated with PGI2 (1 μg/mL) were used as a positive control. Results are mean ± SEM (n ≥ 4). **P < .01; ***P < .001 (calculated by 1-way ANOVA).
62Gap27 modulates PKA activity independently of cAMP. (A) Resting and (B) CRP-XL stimulated (90 seconds) washed human platelets (4 × 108 cells/mL) treated with scrambled peptide (S; 100 µg/mL) or 62Gap27 (50 and 100 μg/mL) for 5 minutes were tested for VASP S157 phosphorylation (a marker of PKA activity). VASP S157 phosphorylation was also evaluated in washed platelets treated with 62Gap27 (100 μg/mL) for 5 minutes in the presence of (C) H89 (10 µM), (D) PKI (10 µM), (F) Rp-8-CPT-cAMPS (200 µM), and (G) SQ 22536 (100 µM). Platelets treated with PGI2 (1 μg/mL) for the stimulation of PKA-mediated phosphorylation were used as positive controls. Sample lysis was carried out using the Laemmli sample buffer before separation by SDS-PAGE. Samples were then transferred to PVDF membranes. 14-3-3-ζ was used as a loading control. (E) Levels of cAMP were measured in resting and CRP-XL (1 μg/mL) stimulated washed human platelets (4 × 108 cells/mL) that had been preincubated with the scrambled peptide (S; 100 µg/mL) or 62Gap27 (50 or 100 µg/mL) for 5 minutes. Platelets treated with PGI2 (1 μg/mL) were used as a positive control. (H-I) Resting washed platelets (4 × 108 cells/mL) from Cx57+/+ and Cx57−/− mice were treated with scrambled peptide (S) or 62Gap27 (100 μg/mL) for 5 minutes in the presence of H89 (10 µM) or Rp-8-CPT-cAMPS (200 µM) and investigated for VASP-S157 phosphorylation. Platelets treated with PGI2 (1 μg/mL) were used as a positive control. Results are mean ± SEM (n ≥ 4). **P < .01; ***P < .001 (calculated by 1-way ANOVA).
VASP phosphorylation was reversed after treatment with PKA inhibitors H89 (10 µM) (Figure 7C) or PKI (10 µM) (Figure 7D), confirming a key role for PKA in this process. We examined cAMP concentration in resting and CRP-XL–activated platelets treated with 62Gap27 (100 µg/mL) or scrambled peptide. Treatment with 62Gap27 did not increase cAMP levels in resting or CRP-XL–stimulated platelets (Figure 7E). Similarly, 62Gap27 did not upregulate cAMP concentration in thrombin-stimulated platelets (supplemental Figure 6C). In agreement with this, 62Gap27-stimulated VASP phosphorylation was not reduced by treatment with Rp-8-CPT-cAMP (200 µM) (Figure 7F), a competitive inhibitor of cAMP binding to PKA or the adenylyl cyclase inhibitor SQ22536 (100 µM) (Figure 7G). Together, these data indicate that 62Gap27 inhibits platelet function through activation of PKA independently of cAMP.
Ser157 VASP phosphorylation was also investigated in Cx57+/+ and Cx57−/− mouse platelets in the presence and absence of PKA inhibitors. Consistent with observations on human platelets, 62Gap27-treated Cx57+/+ mouse platelets exhibited enhanced VASP Ser157 phosphorylation compared with samples treated with scrambled peptide, which was reversed upon treatment with the PKA inhibitor H89 (Figure 7H) but not reversed by Rp-8-CPT-cAMPs (Figure 7I). 62Gap27 treatment of Cx57−/− mouse platelets did not result in VASP phosphorylation, further confirming the specificity of action of 62Gap27 (Figure 7H,I).
It has been reported that PKC isoforms, PI3K, or PKB/Akt can contribute toward the phosphorylation of VASP.32,46,47 However, 62Gap27-induced VASP phosphorylation in platelets was not prevented upon treatment with pan-PKC inhibitor GF109203X (10 µM), PI3K inhibitor LY29400 (100 µM), or AKT inhibitor IV (5 µM) (supplemental Figure 6D-F). Together, these observations provide insight into the mechanism through which the actions of 62Gap27 on Cx62 inhibit platelet activation through the upregulation of PKA activity independently of cAMP.
Discussion
Growing evidence suggests the role of different platelet surface receptors (Eph kinase, CD72, plexin-B1, and CD40L) in contact-dependent signaling that is required for the formation of a stable thrombus.48,49 The contributions of gap junction–mediated intercellular communication to platelet function and thrombus growth represent a recently discovered paradigm for the regulation of circulating cells after cell-cell contact. In this study, we describe the presence of Cx62(57) in platelets, which was found in a punctate arrangement in the cytosol and translocated to the plasma membrane upon activation. These observations are consistent with the formation of hemichannels on the cell membrane and the formation of gap junctions as adjacent platelets make sustained contact within a thrombus. The mechanism by which Cx62 traffics to the cell membrane upon platelet activation remains to be established. In other cell types, connexins translocate to the plasma membrane through the classical secretory pathway.50-54 In platelets, Cx62 is distributed similarly to calreticulin, which is present within the DTS (remnants of megakaryocyte smooth endoplasmic reticulum) but not in subcellular fractions that contain α-granule cargo or cytosolic proteins, suggesting a non-classical secretion mechanism. It has been proposed that connexins are chaperoned by protein disulfide isomerases as connexin extracellular loops are exposed in the endoplasmic reticulum to form disulfide bonds.55 Protein disulfide-isomerases also localize to the platelet DTS and become mobilized in activated platelets, and therefore, they may share mechanisms of trafficking toward the cell surface with Cx62.56
To determine the role of Cx62 in the regulation of platelet function, a novel synthetic mimetic peptide (62Gap27) targeting Cx62(57) was designed. Its inability to inhibit CRP-XL–mediated fibrinogen binding in Cx57−/− mice compared with Cx57+/+ confirmed its selective action toward Cx57/62. Incubation with 62Gap27 significantly downregulated calcein release from activated platelets in suspension (nonaggregated), which points toward a role for connexin hemichannels in the early phases of platelet activation. This was associated with diminished markers of platelet activation such as fibrinogen binding, and dense (ATP release) and α-granule (P-selectin exposure) secretion. It is interesting to note that pannexin-1 releases cytosolic ATP, which primes platelet responses when exposed to low agonist concentrations via the effect of the ATP on P2X1 channels.57 The possibility that direct release of ATP represents a mechanism through which connexins contribute to platelet activation is a priority for investigation in future work. In addition, fluorescence recovery after photobleaching experiments confirmed the formation of Cx62-containing gap junctions between adjacent platelets. Thrombus stability was suppressed both in vitro and in vivo after treatment with 62Gap27, indicating the vital role of gap junction mediated intercellular communication in the reinforcement of thrombi. It is plausible that such robust effects of 62Gap27 on thrombosis in vivo are partly a result of its effects on other circulating or endothelial cells. Further work to explore this will require transgenic mice with platelet-specific deletion of Cx57.
Studies in Cx57-deficient mouse platelets identified that Cx57(62) positively regulates platelet function, hemostasis, and thrombus formation. These functions are shared with Cx37 and Cx40, the other principal platelet connexins.21,26,27,58 We also observed negative regulation of Ca2+ mobilization after treatment with 62Gap27. This inhibition was identified to be partly the result of reduced Ca2+ mobilization from stores, although effects on the Ca2+ influx cannot be ruled out. Numerous channels regulate calcium mobilization in platelets, including P2X1, TRPC6, STIM1, and Orai1.41,42,59 Notably Cx43 has been shown to interact with the calcium channel P2X1.60 It is therefore possible that Cx62 may associate with a platelet calcium channel to influence calcium mobilization.
PKA activation plays an essential role in the regulation of platelet function by controlling several aspects of activation, including integrin αIIbβ3 affinity, inositol 1,4,5-trisphosphate (IP3) receptor function, and shape change via actin polymerization.47,61 Although cAMP is a key activator of PKA in platelets, studies have also reported that PKA activity can be stimulated by collagen or thrombin in a cAMP-independent manner, and in other cells, cAMP-independent PKA activation has been attributed to the effects of sphingosine and free radicals.47,62-64 We provide compelling evidence that the inhibitory effects of 62Gap27 on platelets are mediated through increased PKA activity, independently of cAMP. Notably, cAMP-independent upregulation of PKA signaling occurs in unstimulated platelets, which suggests a direct influence of 62Gap27 binding to Cx62 on PKA activity. The mechanism by which 62Gap27 induces PKA activation remains unclear. Because 62Gap27 is predicted to primarily induce conformational changes in Cx62 (Figure 2C; supplemental Figure 1D), we speculate that 62Gap27 binding may influence the ability of PKA to interact with the connexin or localized A-kinase anchoring proteins that may modulate PKA activity in the vicinity of the connexin. The lack of increased VASP phosphorylation in Cx57-deficient platelets in the absence of 62Gap27 suggests that Cx binding of PKA may result in its activation, while non–Cx57-bound PKA is inactive.
It is possible that connexin-associated PKA may be responsible for the regulation of connexin trafficking (noting that platelet activation results in its recruitment to the plasma membrane) or regulation of channel function. Further work will be required to determine the precise mechanism of PKA activation in the presence or absence of 62Gap27 to assess whether this represents conformational perturbation of Cx62 by the peptide or involves PKA-mediated phosphorylation of the connexin that modulates channel activity. Notably the absence of Cx57, which in itself does not alter PKA-dependent signaling in unstimulated platelets, results in diminished levels of platelet activation. This supports a role for Cx57 channel activity in the function of platelets. It remains to be established whether the inhibitory effects of 62Gap27 are solely mediated through stimulation of PKA signaling (which is dependent on the presence of the connexin) or also through modulation of channel function. It is pertinent that several studies indicate the involvement of PKA and PKG in regulating the phosphorylation of Cx32, Cx35/36, Cx40, Cx43, and Cx50 in various cell types.65-70 In the absence of antibodies that allow the immunoprecipitation of Cx62, we have thus far been unable to determine whether Cx62 represents a PKA substrate in platelets.
The potential link between Cx62 and thrombotic disease risk is uncertain, and relevant mutations that might modulate such risk have not been identified. The expression of Cx62 is not widespread, which may enhance its potential as a therapeutic target, minimizing adverse effects, noting that systemic genetic deletion of Cx57 in mice is well tolerated. Our Gap27 peptide docking experiments are suggestive of regulation of gating by conformational changes although, as discussed, modulation of PKA-dependent signaling may also be important. Non–peptide-based selective inhibitors would need to be developed that potentially mimic peptide binding and/or associated conformational changes to develop this further. A suggested starting point for such work would be to develop biologics that target the proposed 62Gap27 binding sequences predicted in Cx62.
In summary, we have identified key functions for the orphan connexin Cx62(57) in platelets in the regulation of hemostasis and thrombosis. We have revealed a new signaling mechanism through which Cx62(57) and its inhibitor modulate cellular function and highlight the importance of connexin hemichannels and gap junctions in the regulation of the function of circulating cells.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Saudi Cultural Bureau (London, United Kingdom), a Felix Scholarship (2014-17), grants from the British Heart Foundation (RG/09/011/28094 and RG/15/2/31224), the Medical Research Council (MR/J002666/1 and MR/P023878/1), and the Biotechnology and Biological Sciences Research Council.
Authorship
Contribution: K.A.S. and G.D.F. designed the research, performed experiments, analyzed results, and wrote the article; P.S., A.H.M., L.-M.H., S.K.A., and A.E. performed experiments and analyzed results; T.S., A.R.S., R.A., M.A., M.C., N.K., S.V., A.P.B., A.J.U., and C.I.J. performed experiments; and L.J.M. and J.M.G. designed the research, analyzed data, and wrote the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jonathan M. Gibbins, Institute for Cardiovascular and Metabolic Research, School of Biological Sciences, University of Reading, Harborne Building, Whiteknights, Reading RG6 6AS, United Kingdom; e-mail: j.m.gibbins@reading.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal