

Key Points
EVs derived from stored platelets cause TRALI as a function of their elevated ceramide and decreased S1P content.
Inhibiting ceramide formation, supplementing S1P, or washing stored platelets could potentially reduce TRALI incidence and severity.

Abstract
Transfusion-related acute lung injury (TRALI) is a hazardous transfusion complication with an associated mortality of 5% to 15%. We previously showed that stored (5 days) but not fresh platelets (1 day) cause TRALI via ceramide-mediated endothelial barrier dysfunction. As biological ceramides are hydrophobic, extracellular vesicles (EVs) may be required to shuttle these sphingolipids from platelets to endothelial cells. Adding to complexity, EV formation in turn requires ceramide. We hypothesized that ceramide-dependent EV formation from stored platelets and EV-dependent sphingolipid shuttling induces TRALI. EVs formed during storage of murine platelets were enumerated, characterized for sphingolipids, and applied in a murine TRALI model in vivo and for endothelial barrier assessment in vitro. Five-day EVs were more abundant, had higher long-chain ceramide (C16:0, C18:0, C20:0), and lower sphingosine-1-phosphate (S1P) content than 1-day EVs. Transfusion of 5-day, but not 1-day, EVs induced characteristic signs of lung injury in vivo and endothelial barrier disruption in vitro. Inhibition or supplementation of ceramide-forming sphingomyelinase reduced or enhanced the formation of EVs, respectively, but did not alter the injuriousness per individual EV. Barrier failure was attenuated when EVs were abundant in or supplemented with S1P. Stored human platelet 4-day EVs were more numerous compared with 2-day EVs, contained more long-chain ceramide and less S1P, and caused more endothelial cell barrier leak. Hence, platelet-derived EVs become more numerous and more injurious (more long-chain ceramide, less S1P) during storage. Blockade of sphingomyelinase, EV elimination, or supplementation of S1P during platelet storage may present promising strategies for TRALI prevention.
Introduction
Transfusion-related acute lung injury (TRALI), defined as new onset of hypoxemic bilateral lung injury within 6 hours of transfusion of plasma-containing blood products is characterized by lung inflammation and endothelial barrier failure and is associated with high mortality.1-6 TRALI is a global health problem with incidences varying from 1:10000 transfusions to >1:100 in critically ill patients.4,7-10 TRALI is mediated by transfusion of pathogenic donor antibodies and/or biological response modifiers from blood products.11-13 Although mitigation strategies such as deferring female plasma have reduced the incidence of antibody-mediated TRALI, the mechanisms of antibody-independent TRALI remain poorly understood, preventing causal prevention strategies.14
We have previously shown that transfusion of 5-day-stored, but not 1-day-stored, pools of platelets into lipopolysaccharide (LPS)-primed mice causes TRALI.15 In this model, acid sphingomyelinase (ASM), the predominant ceramide-producing enzyme in platelets, was essential for donor platelets to elicit TRALI.15 Our model corroborates human and preclinical data showing ceramide accumulation during platelet storage15-17 and ceramide-induced lung endothelial barrier failure.18,19 Specifically, ceramide causes lung injury when unopposed by sphingosine-1-phosphate (S1P), a counterregulatory sphingolipid that promotes barrier integrity.20 Because S1P decreases while ceramide accumulates in human platelets throughout storage,15 this imbalance in the ratio of ceramide vs S1P (the so-called sphingolipid rheostat21 ) presents an intriguing mechanistic concept for stored platelet-induced TRALI. Biological ceramides are, however, relatively long-chained fatty acids and, as such, are highly hydrophobic, preventing their free circulation in plasma. Hence, the rheostat model necessitates the existence of either a shuttle and/or downstream mechanism for ceramide-mediated lung endothelial barrier failure. Extracellular vesicles (EVs) present a candidate mediator in this scenario as they can serve as lipid shuttles, accumulate during platelet storage,15 and mediate inflammatory processes including lung injury.22 Notably, however, ceramides and EVs may interact in a dual manner in that EVs not only act as hydrophilic chaperones for ceramide, but also require ceramide for their formation. From comprehensive data obtained in a murine TRALI model, an in vitro assay of endothelial barrier function, and analyses of human stored platelets, we present here a new pathogenic concept that identifies stored platelet EVs as novel mediators of TRALI. This effect is tightly controlled by sphingolipids, in that EV biogenesis requires ceramide-forming ASM, whereas EV-induced injury is determined by changes in the sphingolipid rheostat.
Methods
Isolation of EVs from stored platelets
Animal experiments were approved by the Keenan Research Centre (KRC) at St Michael’s Hospital Animal Care Committee. Human platelets were obtained from Canadian Blood Services (CBS; NetCad; Vancouver, BC, Canada) with review ethics board approval from both CBS (2018.002b4r) and KRC (07-207). Human pooled-plasma platelets were stored for 2 to 4 days, platelets from C57BL/6 mice or ASM-deficient (Smpd1−/−) mice for 1 to 5 days (1 day, 3 days, 5 days) as previously described.15,23 Subsets of murine platelets or EVs were treated with the ASM inhibitor ARC3924 (10 mM/L), S1P (50 ng/mL), or with exogenous sphingomyelinase (SM; 0.1 U/mL). EVs were isolated from platelets by differential centrifugation.
EV enumeration and sizing
EVs were diluted (1/100 for murine EVs, 1/1000 for human EVs) in filtered phosphate-buffered saline (PBS) for enumeration and surface abundance of CD41, CD62p, and phosphatidylserine by high-sensitivity flow cytometry (BD FACSARIA III SORP; Becton Dickinson, Franklin Lakes, NJ) and size determination by nanoparticle tracking analysis (NTA) as previously described25,26 (supplemental Figure 1A-B, available on the Blood Web site).
EV sphingolipid characterization
From isolated EVs, S1P, long-chain ceramides (LCCs; C16:0, C18:0, C20:0), and very long chain ceramides (VLCCs; C22:0, C24:0, C24:1) were extracted and quantified by rapid-resolution liquid chromatography and tandem mass spectroscopy as previously described.15,27,28
Lung microvascular endothelial barrier integrity in vitro
Transendothelial electrical resistance (TEER) was measured every 30 minutes in confluent monolayers of primary human pulmonary microvascular endothelial cells (HPMECs) cultured on porous (400 nm) Transwell inserts and treated with EVs, equal volumes of non-EV fractions, or PBS using a REMS Auto Sampler (World Precision Instruments, Sarasota, FL) as described in supplemental Methods. In separate assays, HPMEC Transwells were treated with EVs or PBS and the translocation of 70-kDa fluorescein isothiocyanate (FITC)-dextran (FD70S; Sigma-Aldrich) from the upper to the lower chamber was quantified after 4 hours.
Two-hit murine TRALI model in vivo
Normal saline, murine platelets or EVs were transfused via the tail vein (10 mL/kg) into male 8- to 12-month old BALB/c mice (23-28 g body weight; Charles River Laboratories, Montreal, QC, Canada), which had been primed 2 hours earlier with intraperitoneal 2 mg/kg 0111:B4 Escherichia coli LPS or normal saline as previously described.15 After 6 hours, mice were euthanized under general anesthesia (ketamine/xylazine intraperitoneally). The left lung was lavaged and recovered fluid was assessed for total protein content; the right lung was processed for histological evaluation using a semiquantitative lung injury score (upper lobe), lung tissue myeloperoxidase (MPO) activity (middle lobe), and wet-to-dry lung weight ratio (lower lobe).
Statistical analysis
Statistical analysis was performed using Prism software (version 5.01; GraphPad Software). Data were tested for normal distribution and homoscedasticity, and then analyzed by unpaired (mouse EVs) or paired (human EVs), 2-tailed Student t tests or 1-way analysis of variance followed by the all-pairwise Tukey test. A value of P < .05 was considered statistically significant. Data are depicted as means ± standard deviation (SD).
For methodological details, please see supplemental Methods.
Results
EV formation throughout platelet storage
Throughout storage of murine platelets, the number of EVs in stored platelet supernatants approximately doubled from 1 day to 5 days (Figure 1A). Concomitantly, the size of EVs increased (Figure 1B) and surface abundance of phosphatidylserine decreased, whereas expression of the surface markers CD41 and CD62p remained unchanged (supplemental Figure 2). Previously, we identified ceramide and ASM as critical mediators of antibody-independent TRALI.15 To test whether EVs may serve as biological shuttles for hydrophobic ceramides (C), we assessed EVs from murine platelets stored until 1 day, 3 days, or 5 days (1-, 3-, or 5-day EVs) for LCCs and VLCCs. Relative to 1-day EVs, LCCs were increased in 3- and 5-day EVs, whereas VLCCs were reduced in 5-day EVs as compared with 3-day EVs (Figure 1C).
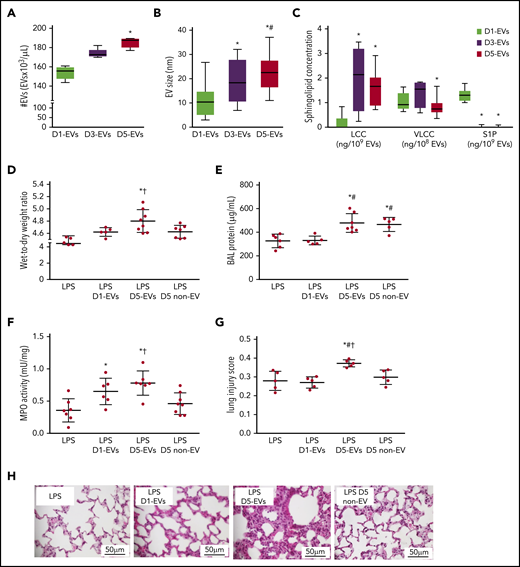
EVs form throughout platelet storage and elicit TRALI in LPS-primed mice. Group data show EV number (#EVs; A), EV size (B), and EV composition in terms of concentrations of LCC and VLCC chain length ceramide and S1P (C) for EVs of platelet pools stored for 1 (D1-EVs), 3 (D3-EVs), or 5 (D5-EVs) days. Wet-to-dry lung weight ratio (D), protein concentration in BAL (E), MPO activity in lung tissue (F), and histological features of lung injury (G) on a quantitative scale from 0 (no injury) to 1 (maximal) were determined in recipient BALB/c male mice 6 hours after transfusion of EVs. Mice were primed for 2 hours with LPS (2 mg/kg intraperitoneally) prior to transfusion of 10 mL/kg body weight of either normal saline (LPS group), EVs of platelet pools stored for 1 (D1-EVs) or 5 (D5-EVs) days, or day 5 platelet- and EV-depleted platelet pools (5 days non-EV). Representative images of hematoxylin-and-eosin–stained histological micrographs show progressive severity of lung injury with transfusion of EVs isolated from platelet pools with increasing storage time in LPS-primed mice. Scale bars, 50 μm (H). Group data for panels A-C are depicted as medians and 25% quartiles; n = 6-15 each, *P < .05 vs D1-EVs and #P < .05 vs D3-EVs (1-way analysis of variance and post hoc all pairwise Tukey test). Group data for panels D-G are given as mean ± SD; n = 5-8 each. *P < .05 vs LPS only, #P < .05 vs LPS plus D1-EVs and †P < .05 vs D5 non-EV (1-way analysis of variance and post hoc all pairwise Tukey test).
EVs form throughout platelet storage and elicit TRALI in LPS-primed mice. Group data show EV number (#EVs; A), EV size (B), and EV composition in terms of concentrations of LCC and VLCC chain length ceramide and S1P (C) for EVs of platelet pools stored for 1 (D1-EVs), 3 (D3-EVs), or 5 (D5-EVs) days. Wet-to-dry lung weight ratio (D), protein concentration in BAL (E), MPO activity in lung tissue (F), and histological features of lung injury (G) on a quantitative scale from 0 (no injury) to 1 (maximal) were determined in recipient BALB/c male mice 6 hours after transfusion of EVs. Mice were primed for 2 hours with LPS (2 mg/kg intraperitoneally) prior to transfusion of 10 mL/kg body weight of either normal saline (LPS group), EVs of platelet pools stored for 1 (D1-EVs) or 5 (D5-EVs) days, or day 5 platelet- and EV-depleted platelet pools (5 days non-EV). Representative images of hematoxylin-and-eosin–stained histological micrographs show progressive severity of lung injury with transfusion of EVs isolated from platelet pools with increasing storage time in LPS-primed mice. Scale bars, 50 μm (H). Group data for panels A-C are depicted as medians and 25% quartiles; n = 6-15 each, *P < .05 vs D1-EVs and #P < .05 vs D3-EVs (1-way analysis of variance and post hoc all pairwise Tukey test). Group data for panels D-G are given as mean ± SD; n = 5-8 each. *P < .05 vs LPS only, #P < .05 vs LPS plus D1-EVs and †P < .05 vs D5 non-EV (1-way analysis of variance and post hoc all pairwise Tukey test).
EVs from stored murine platelets elicit TRALI in LPS-primed mice
To test whether EVs formed during platelet storage may trigger TRALI, we adapted our previously established murine 2-hit TRALI model15 to infuse doses of EVs (without platelets) equivalent to what would be present in 10 mL/kg of transfused pools of platelets into LPS-primed mice. Compared with sham mice receiving only saline after the initial LPS priming, transfused 5-day EVs, but not 1-day EVs replicated the characteristics of murine TRALI in terms of an increased wet-to-dry lung weight ratio (Figure 1D), bronchoalveolar lavage (BAL) fluid total protein concentration (Figure 1E), lung tissue MPO activity (Figure 1F), histological lung injury score (Figure 1G-H) as well as increased serum markers of inflammation (interleukin 1β [IL-1β]; serum amyloid P) (supplemental Figure 3). In line with the 2-hit concept, 5-day EVs in the absence of LPS priming did not induce TRALI (supplemental Figure 4).29 To validate the role of EVs vs soluble mediators in TRALI, we next transfused 5-day platelet pools that had been depleted of both platelets and EVs (5-day non-EV). Five-day non-EV solutions did not cause lung injury relative to LPS controls with the partial exception of BAL protein, which remained elevated (Figure 1D-H).
EVs from stored murine platelets elicit endothelial barrier dysfunction
Next, we assessed the capacity of 1-day vs 5-day EVs to increase endothelial permeability in HPMECs. When HPMECs were administered equal volumes of 1- or 5-day EVs (similar volumetric dosing as for in vivo experiments), a rapid (onset <1 hour) and persistent (>4 hours) loss of endothelial barrier integrity was observed by TEER in response to 5-day EVs only (Figure 2A). Loss of barrier integrity quantified as area-under-the-TEER curve over 4 hours correlated with FITC-dextran translocation in a Transwell assay, consolidating endothelial barrier failure in response to 5-day EVs yet not 1-day EVs (Figure 2B; supplemental Figure 5). Titration of 5-day EVs (1 × 104 to 1 × 106) revealed a dose-dependent progressive loss of endothelial barrier function once 5-day EV numbers exceeded >1 × 105 (supplemental Figure 6). To assess not only quantitative effects of EVs but also qualitative differences, equal numbers (5 × 105) of 1-, 3-, or 5-day EVs were added to HPMECs. At identical doses, 3-and 5-day EVs, but not 1-day EVs disrupted endothelial barrier integrity as measured by TEER (Figure 2C-D), demonstrating that EVs from stored platelets are not only more numerous but also more injurious. Corresponding volume-matched 5 day non-EV solutions did not cause endothelial barrier failure, which, in line with our prior in vivo experiments, confirms EV specificity of this effect (Figure 2C-D).
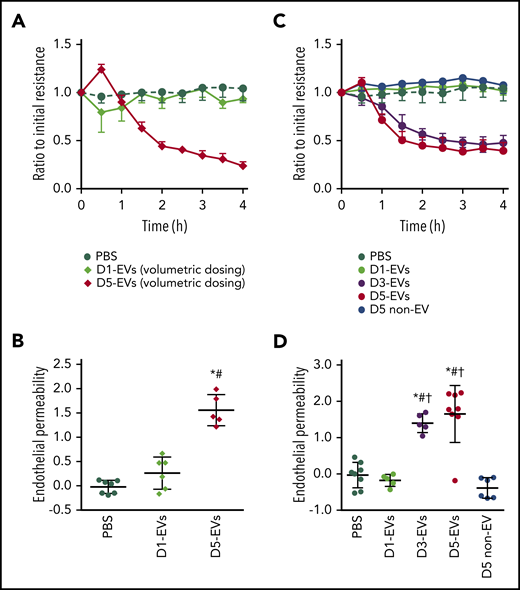
EVs from stored murine platelets elicit endothelial barrier dysfunction. (A) Group data show TEER (measured in ohms) as ratio relative to initial monolayer resistance of primary HPMECs cultured for 4 hours with PBS, EVs from platelets stored for 1 (D1-EVs) or 5 (D5-EVs) days. EVs were dosed volumetrically (diamonds) using a similar strategy as in the in vivo experiments. (B) For each individual TEER measurement, the area under the curve (AUC) relative to the baseline y = 1.0 was calculated and is given as group data for endothelial permeability, with increased AUC indicating loss of resistance (increased permeability), whereas 0 or negative values indicate maintenance of or increased endothelial barrier function. (C) Group data show TEER in HPMEC monolayers cultured with PBS, or equal numbers (circles; 5 × 105 EVs each instead of volumetric dosing as seen in panels A-B) of D1-EVs, D3-EVs, or D5-EVs. Day 5 platelet- and EV-depleted platelet pools (D5 non-EV) were administered in identical volumes (5 µL) as D5-EVs Corresponding calculated AUCs are given in panel D. Group data are depicted as mean ± SD; n = 5-8 each. *P < .05 vs PBS only, #P < .05 vs D1-EVs and †P < .05 vs D5 non-EV (1-way analysis of variance and post hoc all pairwise Tukey test).
EVs from stored murine platelets elicit endothelial barrier dysfunction. (A) Group data show TEER (measured in ohms) as ratio relative to initial monolayer resistance of primary HPMECs cultured for 4 hours with PBS, EVs from platelets stored for 1 (D1-EVs) or 5 (D5-EVs) days. EVs were dosed volumetrically (diamonds) using a similar strategy as in the in vivo experiments. (B) For each individual TEER measurement, the area under the curve (AUC) relative to the baseline y = 1.0 was calculated and is given as group data for endothelial permeability, with increased AUC indicating loss of resistance (increased permeability), whereas 0 or negative values indicate maintenance of or increased endothelial barrier function. (C) Group data show TEER in HPMEC monolayers cultured with PBS, or equal numbers (circles; 5 × 105 EVs each instead of volumetric dosing as seen in panels A-B) of D1-EVs, D3-EVs, or D5-EVs. Day 5 platelet- and EV-depleted platelet pools (D5 non-EV) were administered in identical volumes (5 µL) as D5-EVs Corresponding calculated AUCs are given in panel D. Group data are depicted as mean ± SD; n = 5-8 each. *P < .05 vs PBS only, #P < .05 vs D1-EVs and †P < .05 vs D5 non-EV (1-way analysis of variance and post hoc all pairwise Tukey test).
EV formation throughout storage of murine platelets requires ASM
Formation of EVs is considered to rely, at least in part, on formation of ceramide,30 which in platelets is predominantly produced from sphingomyelin by ASM.31 Considering 5-day EVs had greater LCC content and were more injurious than 1-day EVs, we next assessed the role of ASM in EV formation and TRALI induction. Stored platelets from ASM-deficient (Smpd1−/−) donor mice or wild-type (WT) platelets stored in the presence of the ASM inhibitor ARC39 both showed reduced formation of EVs over 5 days of storage as compared with untreated WT platelets (Figure 3A). Conversely, addition of exogenous SM to WT platelets during storage increased the number of EVs formed after only 1 day of storage in comparison with platelets stored without SM (Figure 3B). Notably, although inhibition of ASM during platelet storage attenuated the increase in EV size over storage time, supplementation with SM did not increase EV size. These findings indicate that ceramide can directly trigger EV formation; in contrast, it is required but by itself not sufficient to increase EV size (Figure 3C-D). Addition of exogenous SM to platelets for 24 hours increased EV LCCs as compared with untreated WT 1-day EVs (Figure 3E). However, ASM deficiency or inhibition did not reduce EV ceramide content, suggesting that EV formation from stored platelets requires a minimal level of EV associated ceramide species. Unexpectedly, EVs from 5-day Smpd1−/− or ARC39-treated WT platelets had in fact higher levels of LCCs and VLCCs as compared with 5-day EVs from stored WT platelets (Figure 3F).
![EV formation throughout storage of murine platelets requires ASM. Group data show EV number (#EVs; A-B), EV size (C-D), and EV composition in terms of concentrations of LCC and VLCC chain length ceramide and S1P (E-F) for EVs from WT platelet pools stored for 1 (D1-EVs) or 5 days (D5-EVs) with or without addition of 10 μM/L of the ASM inhibitor ARC39 (D5-EVs [ARC39]) or 0.1 U/mL SM (D1-EVs [SM]), or EVs from 5 days stored platelets from Smpd1−/− mice (D5-EVs [Smpd1−/−]). Group data are depicted as medians and 25% quartiles; n = 6-18 each. *P < .05 vs D5-EVs (A,C,F) or D1-EVs (B,D,E), respectively (1-way analysis of variance and post hoc all pairwise Tukey test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/5/10.1182_blood.2020005985/1/m_bloodbld2020005985f3.png?Expires=1767590775&Signature=nCMvYhbT6tLkZnZLCA5q-mWOF2w6MTh7vlKxwr-Zkj7lwWNli~jKdVhjWmHPXLKuAwCf7tGxE9Jhc~ngTd0xXCGkoIy7feo1XT7A8M0KlljV1v3SkJ1ooG9CnnMzMBK4NQ6bsd7iZQnlDj9i5NBbVhyFwZCQl4zZlyuPUWA~fAur4clp4E9Yb2e1XMcWl~HJszS4FFTlfrg7sjYeO4PMsLRh2dm-bnFMN8R~dl9SVluCqCNA82MWz86xrDx9~HXBXaM0dLM8chmFlBmwddHpwlszlcgInsSXjDmjl47HP89XOCHjK9RdV3cmLPSpGmzCEgCiiVTBJEjCJm4~qsv1NA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
EV formation throughout storage of murine platelets requires ASM. Group data show EV number (#EVs; A-B), EV size (C-D), and EV composition in terms of concentrations of LCC and VLCC chain length ceramide and S1P (E-F) for EVs from WT platelet pools stored for 1 (D1-EVs) or 5 days (D5-EVs) with or without addition of 10 μM/L of the ASM inhibitor ARC39 (D5-EVs [ARC39]) or 0.1 U/mL SM (D1-EVs [SM]), or EVs from 5 days stored platelets from Smpd1−/− mice (D5-EVs [Smpd1−/−]). Group data are depicted as medians and 25% quartiles; n = 6-18 each. *P < .05 vs D5-EVs (A,C,F) or D1-EVs (B,D,E), respectively (1-way analysis of variance and post hoc all pairwise Tukey test).
EV formation throughout storage of murine platelets requires ASM. Group data show EV number (#EVs; A-B), EV size (C-D), and EV composition in terms of concentrations of LCC and VLCC chain length ceramide and S1P (E-F) for EVs from WT platelet pools stored for 1 (D1-EVs) or 5 days (D5-EVs) with or without addition of 10 μM/L of the ASM inhibitor ARC39 (D5-EVs [ARC39]) or 0.1 U/mL SM (D1-EVs [SM]), or EVs from 5 days stored platelets from Smpd1−/− mice (D5-EVs [Smpd1−/−]). Group data are depicted as medians and 25% quartiles; n = 6-18 each. *P < .05 vs D5-EVs (A,C,F) or D1-EVs (B,D,E), respectively (1-way analysis of variance and post hoc all pairwise Tukey test).
EVs from stored murine Smpd1−/− platelets cause less TRALI in LPS-primed mice
To test whether 5-day EVs from Smpd1−/− platelets (5-day EVs [Smpd1−/−]) cause less TRALI as compared with 5-day EVs from WT platelets (5-day EVs (WT)), we next transfused 10 mL/kg (volumetric dosing) 5-day EVs from platelet pools of either WT or Smpd1−/− mice into LPS-primed mice. Characteristic aspects of TRALI as seen with transfusion of 5-day EVs (WT) were significantly attenuated after transfusion of 5-day EVs (Smpd1−/−), as demonstrated by lower wet-to-dry lung weight ratios (Figure 4A), BAL total protein concentration (Figure 4B), and histological lung injury scores (Figure 4C-E). MPO activity in lung tissue was also lower following transfusion of 5-day EVs (Smpd1−/−), yet without reaching statistical significance. Importantly, these findings identify ASM as promising pharmacological target to reduce the injuriousness of stored platelets.
![EVs from stored murine Smpd1−/−platelets cause less TRALI in LPS-primed mice. Group data show wet-to-dry lung weight ratio (A), BAL protein concentration (B), MPO activity in lung tissue (C), and histological features of lung injury (D) on a quantitative scale from 0 (no injury) to 1 (maximal) in recipient BALB/c male mice (6 hours after transfusion). Mice were primed for 2 hours with LPS (2 mg/kg intraperitoneally) prior to 10 mL/kg body weight transfusion of either EVs obtained from WT (D5-EVs [WT]) or Smpd1−/− (D5-EVs [Smpd−/−]) murine platelet pools after 5 days storage. Representative images of hematoxylin-and-eosin–stained histological micrographs (scale bars, 50 μm) show higher severity of lung injury following transfusion of D5-EVs (WT) compared with D5-EVs (Smpd1−/−) (E). Group data are depicted as mean ± SD; n = 5-8 each. *P < .05 vs LPS 5-day EVs (WT) (2-tailed Student t test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/5/10.1182_blood.2020005985/1/m_bloodbld2020005985f4.png?Expires=1767590775&Signature=Q1uvzQ-dkOIIhyiMaxoTxBy1yHuqMMn1uvYAC4KBVHb5CkatqiTCznjbHzeiXoGlFXp4IMLyi92jFtxzClFkAff1YXdD4oaRvEbvkzEhZL~OcMhPOQgdRPZ1hf-UGQRqDx-vlH7~inQ8Re8DisQr5C5jrSu3423CYfmlQWzgdR1CzNew~X9IlSiQv3uepSc2bJz4jq1Ode4mx1mQP2aa3AsQh2~V9INQ~ObJbXrHTBIPPAJNpTm2NrdsASk~s2A5~YBCvmyp-2MqElJw8FL3e8~8cjv9HQwxexQEIfDSJ0fI9W9rvQixg1WXH7zSJdxXFA1-lXu-EJyn2RayA~SA1g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
EVs from stored murine Smpd1−/−platelets cause less TRALI in LPS-primed mice. Group data show wet-to-dry lung weight ratio (A), BAL protein concentration (B), MPO activity in lung tissue (C), and histological features of lung injury (D) on a quantitative scale from 0 (no injury) to 1 (maximal) in recipient BALB/c male mice (6 hours after transfusion). Mice were primed for 2 hours with LPS (2 mg/kg intraperitoneally) prior to 10 mL/kg body weight transfusion of either EVs obtained from WT (D5-EVs [WT]) or Smpd1−/− (D5-EVs [Smpd−/−]) murine platelet pools after 5 days storage. Representative images of hematoxylin-and-eosin–stained histological micrographs (scale bars, 50 μm) show higher severity of lung injury following transfusion of D5-EVs (WT) compared with D5-EVs (Smpd1−/−) (E). Group data are depicted as mean ± SD; n = 5-8 each. *P < .05 vs LPS 5-day EVs (WT) (2-tailed Student t test).
EVs from stored murine Smpd1−/−platelets cause less TRALI in LPS-primed mice. Group data show wet-to-dry lung weight ratio (A), BAL protein concentration (B), MPO activity in lung tissue (C), and histological features of lung injury (D) on a quantitative scale from 0 (no injury) to 1 (maximal) in recipient BALB/c male mice (6 hours after transfusion). Mice were primed for 2 hours with LPS (2 mg/kg intraperitoneally) prior to 10 mL/kg body weight transfusion of either EVs obtained from WT (D5-EVs [WT]) or Smpd1−/− (D5-EVs [Smpd−/−]) murine platelet pools after 5 days storage. Representative images of hematoxylin-and-eosin–stained histological micrographs (scale bars, 50 μm) show higher severity of lung injury following transfusion of D5-EVs (WT) compared with D5-EVs (Smpd1−/−) (E). Group data are depicted as mean ± SD; n = 5-8 each. *P < .05 vs LPS 5-day EVs (WT) (2-tailed Student t test).
EVs from stored murine platelets are equally injurious independent of ASM activity
In line with the in vivo finding that transfusion of equal volumes of 5-day EVs (Smpd1−/−) causes less injury as compared with 5-day EVs (WT) in mice, volumetric dosing of 5-day EVs (Smpd1−/−) caused no endothelial barrier failure in vitro, yet injury in response to volumetric EV dosing was restored when 5-day (Smpd1−/−) platelets were supplemented with exogenous SM throughout storage (Figure 5A). These findings may be attributable not only to a lesser (volumetric) abundance of EVs in the absence of ASM but potentially also to a reduced injuriousness per individual EV. To assess potential qualitative differences between EVs, in vitro HPMEC barrier analyses were conducted with matched EV numbers instead of volumetrically matched doses of EVs from stored platelets. Addition of 5 × 105 5-day EVs produced similar barrier failure independent of the presence (WT), deficiency (Smpd1−/−), or pharmacological inhibition (ARC39) of ASM (Figure 5B-C). This result is in line with the well-established barrier-disruptive effect of ceramide in lung injury32 and our previous finding that ASM inhibition or deficiency reduced EV abundance, but not individual EV ceramide content. Surprisingly, however, EVs from SM-treated 1 day platelets with elevated ceramide levels (Figure 3E) did not increase endothelial permeability as compared with untreated 1-day EVs (Figure 5B-C). Hence, the presence of ceramide in stored platelet EVs is required, but not sufficient to cause lung endothelial barrier failure, implicating another factor beyond ceramide.
![Role of ceramide and S1P in endothelial barrier regulation by murine platelet EVs. (A) Group data show calculated AUC for TEER relative to initial monolayer resistance in HPMECs cultured for 4 hours with equal volumes of EVs attained from 5-day (Smpd1−/−) platelets treated throughout storage (on day 0, 2, and 4) with 0.1 U/mL SM or with equivalent volume of normal saline. (B-C) Group data show TEER relative to initial monolayer resistance in HPMECs cultured for 4 hours with PBS, EVs from 1 day of storage with (D1-EVs [SM]) or without (D1-EVs [WT]) addition of 0.1 U/mL sphingomyelinase (SM), or EVs from 5 days of platelet storage with (D5-EVs [ARC39]) or without (D5-EVs [WT]) 10 μM/L of the ASM inhibitor ARC39, or EVs from 5 days of storage of Smpd1−/− platelets (D5-EVs [Smpd1−/−]) with equal numbers of EVs (circles; 5 × 105 EVs each) (B), and corresponding calculated AUCs (C). (D-E) Group data show TEER in HPMECs cultured with PBS, with 50 ng/mL S1P, or EVs from 5 days stored platelets with (D5-EVs [S1P]) or without (D5-EVs) 50 ng/mL S1P with equal numbers of EVs (circles each; 5 × 105 EVs) (D), and corresponding calculated AUCs (E). Group data are depicted as mean ± SD; n = 5-8 each. *P < .05 vs (D5-EVs [Smpd1−/−]) (A); *P < .05 vs PBS, #P < .05 vs D1-EVs (WT) (C); and *P < .05 vs PBS, #P < .05 D5-EVs (S1P) (E) (1-way analysis of variance and post hoc all pairwise Tukey test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/5/10.1182_blood.2020005985/1/m_bloodbld2020005985f5.png?Expires=1767590775&Signature=Yj1FGkfckEsrmaYK9Jwe6XzqFgQZA5esWavqVIUdfDqyeN5mZgQdQFON5P5XI4TYj6UB1ILsr1RVaxaeHNsC6F-5I8QnmgT77GJhEET8EEBaaL6RQEv9gewHMkdJaaHB7vOkuuVqMJPaC9H9Z9T5Z6miXI00m1MX-fv2SqkoDOxr9i6VSgwB7-apNOz7y-A0iRr40uj1KELs5DSwt0eVTL1QEJE-zkra6ZRzpWGuTRTsuTiMln5KAnQizCPW1cy8nLocR17kMxYO9crguKzFfBhNXqmiQo8iJWYx7oDhnn6v6JxY1lleANwFzckTKLJude1VcnmMkQx7TqEVYhHDSA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Role of ceramide and S1P in endothelial barrier regulation by murine platelet EVs. (A) Group data show calculated AUC for TEER relative to initial monolayer resistance in HPMECs cultured for 4 hours with equal volumes of EVs attained from 5-day (Smpd1−/−) platelets treated throughout storage (on day 0, 2, and 4) with 0.1 U/mL SM or with equivalent volume of normal saline. (B-C) Group data show TEER relative to initial monolayer resistance in HPMECs cultured for 4 hours with PBS, EVs from 1 day of storage with (D1-EVs [SM]) or without (D1-EVs [WT]) addition of 0.1 U/mL sphingomyelinase (SM), or EVs from 5 days of platelet storage with (D5-EVs [ARC39]) or without (D5-EVs [WT]) 10 μM/L of the ASM inhibitor ARC39, or EVs from 5 days of storage of Smpd1−/− platelets (D5-EVs [Smpd1−/−]) with equal numbers of EVs (circles; 5 × 105 EVs each) (B), and corresponding calculated AUCs (C). (D-E) Group data show TEER in HPMECs cultured with PBS, with 50 ng/mL S1P, or EVs from 5 days stored platelets with (D5-EVs [S1P]) or without (D5-EVs) 50 ng/mL S1P with equal numbers of EVs (circles each; 5 × 105 EVs) (D), and corresponding calculated AUCs (E). Group data are depicted as mean ± SD; n = 5-8 each. *P < .05 vs (D5-EVs [Smpd1−/−]) (A); *P < .05 vs PBS, #P < .05 vs D1-EVs (WT) (C); and *P < .05 vs PBS, #P < .05 D5-EVs (S1P) (E) (1-way analysis of variance and post hoc all pairwise Tukey test).
Role of ceramide and S1P in endothelial barrier regulation by murine platelet EVs. (A) Group data show calculated AUC for TEER relative to initial monolayer resistance in HPMECs cultured for 4 hours with equal volumes of EVs attained from 5-day (Smpd1−/−) platelets treated throughout storage (on day 0, 2, and 4) with 0.1 U/mL SM or with equivalent volume of normal saline. (B-C) Group data show TEER relative to initial monolayer resistance in HPMECs cultured for 4 hours with PBS, EVs from 1 day of storage with (D1-EVs [SM]) or without (D1-EVs [WT]) addition of 0.1 U/mL sphingomyelinase (SM), or EVs from 5 days of platelet storage with (D5-EVs [ARC39]) or without (D5-EVs [WT]) 10 μM/L of the ASM inhibitor ARC39, or EVs from 5 days of storage of Smpd1−/− platelets (D5-EVs [Smpd1−/−]) with equal numbers of EVs (circles; 5 × 105 EVs each) (B), and corresponding calculated AUCs (C). (D-E) Group data show TEER in HPMECs cultured with PBS, with 50 ng/mL S1P, or EVs from 5 days stored platelets with (D5-EVs [S1P]) or without (D5-EVs) 50 ng/mL S1P with equal numbers of EVs (circles each; 5 × 105 EVs) (D), and corresponding calculated AUCs (E). Group data are depicted as mean ± SD; n = 5-8 each. *P < .05 vs (D5-EVs [Smpd1−/−]) (A); *P < .05 vs PBS, #P < .05 vs D1-EVs (WT) (C); and *P < .05 vs PBS, #P < .05 D5-EVs (S1P) (E) (1-way analysis of variance and post hoc all pairwise Tukey test).
S1P content modulates the injuriousness of EVs
In tissue homeostasis, the effects of ceramide are often opposed by another sphingolipid, S1P, which together form a regulatory sphingolipid rheostat.21 In the pulmonary circulation, S1P acts as a barrier stabilizer that counters the harmful effect of ceramide on barrier integrity.33 In previous work, we and others have shown that 5-day stored platelets not only gain ceramide but concomitantly lose S1P.15 To test whether this loss of S1P also extends to platelet-derived EVs, and may contribute to the barrier-disruptive effects of EVs from stored platelets, we analyzed EV-S1P content throughout platelet storage. As compared with 1-day EVs, 3-day and 5-day EVs showed an almost complete loss in S1P, which was not affected by ASM deficiency (Smpd1−/−) or inhibition (Arc39) (Figures 1C and 3F). This loss of S1P may increase the potential of EVs to induce lung injury, which could explain why 1-day EVs from SM-treated platelets (with elevated ceramide but preserved S1P) do not cause injury. To test this notion, we probed whether endothelial barrier failure in response to 5-day EVs may be prevented by restoration of S1P levels. Indeed, addition of exogenous S1P to murine platelets during storage completely prevented HPMEC barrier loss in response to 5-day EVs (Figure 5D-E). Similarly, addition of exogenous S1P to 5-day platelets prior to transfusion into LPS-primed mice reduced the severity of TRALI (supplemental Figure 7).
Stored human platelets replicate increased EV formation, altered EV sphingolipid rheostat, and EV-mediated endothelial barrier failure
To assess the translational relevance of our findings for human disease, we characterized EV formation, sphingolipid profiles, and endothelial injury potential of EVs from human platelets stored for 2 or 4 days. Four-day EVs were more numerous (Figure 6A), and had greater LCC, lower VLCC, and lower S1P content (Figure 6C-E) compared with 2-day EVs. Unlike murine EVs, however, human EVs did not show a consistent increase in size throughout storage (Figure 6B). Importantly, equal volumetric doses of 4-day EVs, yet not 2-day EVs caused rapid (onset <1 hour) loss of endothelial barrier integrity that persisted for >4 hours, thus replicating the injurious effect of EVs from stored murine platelets (Figure 6F).
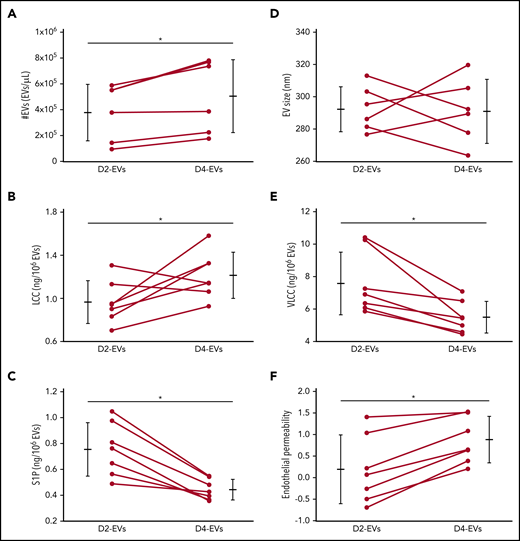
Stored human platelets replicate increased EV formation, altered EV sphingolipid rheostat, and EV-mediated endothelial barrier failure. Group data show comparisons between EVs from human platelets stored for either 2 (D2-EVs) or 4 (D4-EVs) days in terms of EV number (#EVs) (A), EV size (B), EV composition in terms of concentrations of LCC (C), VLCC (D), and S1P (E), and the calculated AUC for TEER relative to initial monolayer resistance represented as endothelial permeability (F) from TEER data determined after treatment of HPMEC monolayers with volumetric doses of EVs. Data are depicted as mean plus or minus SD) for group data (outer bars) as well as individual data points with lines connecting values from the same sample at both time points, n = 5-7 each, *P < .05 vs D2-EVs (paired, 2-tailed Student t test).
Stored human platelets replicate increased EV formation, altered EV sphingolipid rheostat, and EV-mediated endothelial barrier failure. Group data show comparisons between EVs from human platelets stored for either 2 (D2-EVs) or 4 (D4-EVs) days in terms of EV number (#EVs) (A), EV size (B), EV composition in terms of concentrations of LCC (C), VLCC (D), and S1P (E), and the calculated AUC for TEER relative to initial monolayer resistance represented as endothelial permeability (F) from TEER data determined after treatment of HPMEC monolayers with volumetric doses of EVs. Data are depicted as mean plus or minus SD) for group data (outer bars) as well as individual data points with lines connecting values from the same sample at both time points, n = 5-7 each, *P < .05 vs D2-EVs (paired, 2-tailed Student t test).
Discussion
Here, we report a novel sphingolipid- and EV-mediated pathomechanism for platelet-mediated TRALI. Specifically, ceramide first promotes formation of EVs, which then deliver ceramide to the lung where it causes endothelial barrier failure when the EV-sphingolipid rheostat is further imbalanced by low S1P levels (Figure 7A). During storage of human or mouse platelets, EVs became more abundant and were increasingly able to elicit endothelial monolayer barrier leak in vitro and, for murine platelet-derived EVs, induce characteristic symptoms of TRALI in vivo. These barrier-disruptive effects were attributable to progressive ceramide accumulation in the platelet pools,15 as inhibition or deficiency of ASM throughout 5 days of platelet storage decreased EV abundance and their ability to induce lung damage. The latter effect was largely attributable to EVs being less numerous, as the injury potential for equal numbers of 5-day EVs was unaffected by ASM inhibition or deficiency (Figure 7B). Conversely, although supplementation of platelets with SM for 24 hours increased EV abundance and ceramide content, it did not suffice for EVs to cause endothelial barrier failure in vitro (Figure 7C), presumably because these EVs still contain sufficient levels of S1P to counteract the effects of ceramide. Corroborating the relevance of S1P in barrier regulation, supplementation of 5-day EVs with S1P restored barrier function in vitro and supplementation of 5-day platelets with S1P reduced lung injury in vivo (Figure 7D). Taken together, these findings identify platelet-derived EVs as novel mediators of (and thus, potential therapeutic targets in) TRALI and may explain why the incidence and severity of TRALI increases with the duration of storage time.
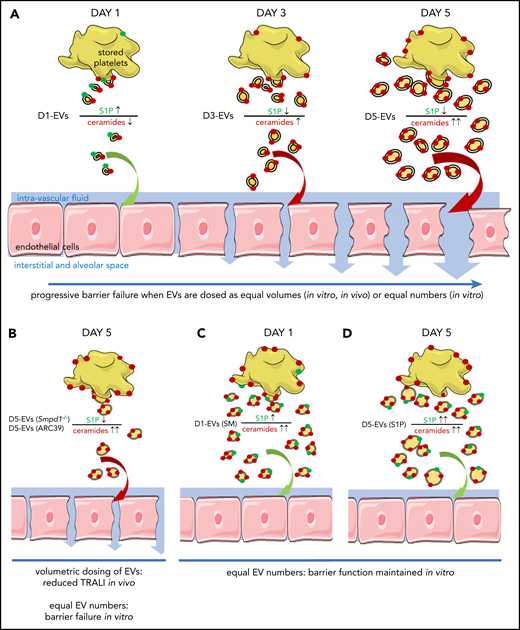
Schematic concept how EVs and imbalances of the sphingolipid rheostat conjointly cause endothelial barrier failure. (A) As platelets are stored over time, they form increasing amounts of EVs which become larger, enriched in long chain ceramides, and S1P deplete. This abundance in EVs with an imbalanced sphingolipid rheostat leads to endothelial barrier failure in vitro and in vivo. (B) Inhibition of ASM by pharmacological blockade (10 μg/mL ARC39) or genetic absence (Smpd1−/−) of ASM leads to less numerous but similarly ceramide enriched and S1P deplete D5-EVs after 5 days of platelet storage. Volumetric transfusion of these EVs causes less TRALI in vivo due to lower abundance, although equal doses of these EVs still cause barrier failure in vitro due to an imbalanced sphingolipid rheostat. (C) The presence of elevated EV-ceramide alone is insufficient to cause barrier failure in vitro, as platelets stored for 1 day with SM release ceramide-enriched but also S1P-rich EVs, which fail to cause barrier failure when dosed in equal numbers in vitro. (D) Corroborating that the presence of S1P can prevent barrier failure by LCC-rich EVs, supplementation of S1P prevented barrier failure in response to D5-EVs.
Schematic concept how EVs and imbalances of the sphingolipid rheostat conjointly cause endothelial barrier failure. (A) As platelets are stored over time, they form increasing amounts of EVs which become larger, enriched in long chain ceramides, and S1P deplete. This abundance in EVs with an imbalanced sphingolipid rheostat leads to endothelial barrier failure in vitro and in vivo. (B) Inhibition of ASM by pharmacological blockade (10 μg/mL ARC39) or genetic absence (Smpd1−/−) of ASM leads to less numerous but similarly ceramide enriched and S1P deplete D5-EVs after 5 days of platelet storage. Volumetric transfusion of these EVs causes less TRALI in vivo due to lower abundance, although equal doses of these EVs still cause barrier failure in vitro due to an imbalanced sphingolipid rheostat. (C) The presence of elevated EV-ceramide alone is insufficient to cause barrier failure in vitro, as platelets stored for 1 day with SM release ceramide-enriched but also S1P-rich EVs, which fail to cause barrier failure when dosed in equal numbers in vitro. (D) Corroborating that the presence of S1P can prevent barrier failure by LCC-rich EVs, supplementation of S1P prevented barrier failure in response to D5-EVs.
Stored platelets continue to have a very high per-product-risk for TRALI even in an era of antibody mitigation.4,34,35 Here, we show that both human and murine platelets produce increasing numbers of EVs during storage, consistent with previous findings in stored human platelets.16 EVs are small (<1 μm diameter) vesicles confined by a lipid bilayer which can be released spontaneously, but are formed in greater numbers and frequently with altered cargo following cell stress or activation.29,36 Due to their ability to transport and deliver genomic, proteomic, or lipidomic material, EVs in the blood stream can serve as shuttles that regulate homeostatic or pathological processes.22 Of late, EVs have been proposed to contribute, at least in part, to transfusion related adverse events and in particular to transfusion-related immunomodulation.22,37-39 Using our previously established murine model of antibody-independent TRALI,15 we now provide proof-of-principle for the critical contribution of EVs to the induction of TRALI by stored platelets. As compared with 1-day EVs, 5-day EVs induced characteristic signs of TRALI including lung edema formation, protein extravasation, and histological evidence of lung injury. These effects were attributable to EVs rather than non-EV soluble mediators, as EV-depleted supernatants from 5-day platelets did not have a similarly injurious potential. These findings support clinical data identifying EVs as potential biomarkers of TRALI40,41 and identify their critical pathophysiologic role in the progressive increase of TRALI risk throughout storage. Mechanistically, increasing TRALI severity as a function of platelet storage time could be linked to the release of more numerous and more injurious EVs causing a progressive loss of endothelial barrier function, a characteristic hallmark of TRALI. Notably, the detrimental effect of 5-day EVs on endothelial barrier function in vitro in the absence of immune cells indicates that EVs can trigger TRALI by exerting direct injurious effects on the alveolo-capillary barrier. The observed proinflammatory effects with lung neutrophil accumulation and elevated systemic levels of IL-1β and serum amyloid P further suggest that EV-induced immunomodulation (as in transfusion-related immunomodulation) may act synergistically with such direct effects. Specifically, coculture studies demonstrated that platelet EVs can activate neutrophils to aggravate barrier failure by causing endothelial cell death,42,43 thus complementing the characteristic features of acute lung injury, namely barrier failure and hyperinflammation.
Previously, we found ceramide, the metabolic product of ASM, to accumulate in platelets throughout storage in an ASM-dependent manner and demonstrated a critical role for platelet ASM in the induction of TRALI following transfusion of 5-day platelets.15 Here, we provide a mechanistic explanation for these findings, in that we found the progressive release of injurious EVs during platelet storage to be dependent on ASM, as EV formation was reduced in both WT platelets stored in the presence of the ASM inhibitor ARC39 or in Smpd1−/− platelets. This finding is consistent with previous reports linking ceramide and ASM to the formation of EVs.30,44-48 Specifically, blockade of ASM has been shown to reduce EV formation in glial and red blood cells.30,48 We now provide similar data for platelets, in which ASM inhibition or deficiency prevents the storage-time-dependent accumulation of ceramide,15 and accordingly, EV release. Interestingly, inhibition or deficiency of ASM during 5 days of platelet storage reduced EV abundance but not ceramide content per individual EVs. This finding may suggest that, in keeping with ceramide’s role in EV biogenesis,46,49 a certain ceramide content is required for EV formation during platelet storage. Why this threshold in ceramide content is seemingly higher for 3-day EVs and 5-day EVs as compared with 1-day EVs remains unclear, but may point to essential differences in the cellular mechanisms driving basal vs storage-time-dependent platelet-EV release. Counterintuitively, ASM inhibition or deficiency, albeit reducing ceramide levels in stored platelets,15 increased the content of LCCs and VLCCs per EV, suggesting preferential ceramide packaging into EVs under conditions of relative ceramide deficiency. The molecular basis and physiological relevance of this effect remain presently unclear, but could point toward novel feedback loops in the regulation of cellular and EV ceramide content.
In addition to enhancing the formation of injurious EVs, ceramide may also directly participate in EV-mediated lung injury as it is enriched in EVs50 and constitutes a key mediator in acute lung injury.15,51 Importantly, the notion of ceramide delivery to target cells via EVs solves a central conceptual dilemma in our understanding of how sphingolipids disseminate homeostatic and disease signals. Biological ceramides are long-chain lipids and, as such, are poorly soluble in water or plasma.52 Although their detrimental role in acute lung injury and related diseases such as sepsis is well established,53 the actual mechanism of their shuttling between cells and within the circulation remains unclear. Here, we show that EVs act as hydrophilic chaperones that facilitate the transport of ceramide in plasma by both ceramide enrichment of EVs and increased EV abundance, ultimately triggering TRALI by a dual mechanism, namely a higher number of more injurious EVs. Yet, increased transport of LCCs alone cannot sufficiently explain the onset of TRALI, as SM supplementation increased 1-day EV ceramide content and numbers to values equal to 5-day EVs, yet without causing endothelial barrier failure in vitro. This seemingly discordant finding may be explained by the sphingolipid rheostat, that is, the fact that ceramide’s proapoptotic and barrier-disruptive properties are at least partially counteracted by S1P.33 In keeping with his concept, even EVs with high ceramide content failed to elicit barrier failure provided that S1P levels remained normal or were substituted, as seen in 1-day EVs from SM-treated platelets or in 5-day EVs supplemented with S1P. As such, changes in sphingolipids elicit antibody-independent TRALI via a dual mechanism: (1) ceramide increases the number of EVs released during platelet storage, which dose-dependently cause endothelial barrier failure, due to (2) an altered sphingolipid composition with increased LCC content in the absence of the counterbalancing effects of S1P.
Notably, recent findings suggest differential functional roles of ceramide species dependent on the chain length of the sphingoid base, in that LCCs have been proven injurious in models of acute lung injury,54,55 whereas VLCCs can exert protective effects.54-56 One may thus speculate that the higher injuriousness of prolonged (4-5 day) storage EVs as compared with EVs from platelets stored 1 to 2 days may not only be attributable to a higher content of LCCs (and a loss of S1P), but also to a relative paucity in VLCCs. The functional relevance of this putative second rheostat is, however, less clear and remains to be elucidated in greater depth.
Several limitations are relevant for the interpretation of our findings. First, a mouse model was used for functional proof-of-concept, which is not feasible in human trials, even though key storage-time-dependent characteristics such as enhanced EV release and altered sphingolipid composition could be replicated in human platelets. A second limitation is that we tested pooled plasma platelets and as such cannot directly extrapolate findings to other clinical platelet products. Finally, it should be noted that we do not provide direct proof for the notion that the higher LCC content directly contributes to the higher injuriousness of 5-day EVs. Elevated LCC levels are a characteristic (and presumably, essential) feature of 5-day EVs that is unaltered by ASM-targeted interventions, hence preventing testing of 5-day EVs low in ceramide. Yet, in view of the well-documented barrier-disruptive effects of ceramide and its counterregulation by S1P, we consider our interpretation of an imbalanced sphingolipid rheostat plausible.
We show that throughout storage, platelets release increasing numbers of EVs in an ASM-dependent manner that bear progressively greater injurious potential due to a cumulative shift of the sphingolipid rheostat from S1P to ceramide. As such, both ASM and EVs emerge as novel therapeutic targets in stored platelets. EV levels within stored human blood products may be monitored as biomarker to assess safety and guide mitigation strategies such as platelet washing to reduce injurious EVs.57 Pharmacological inhibition of ASM presents an attractive strategy, as such intervention could be limited to stored platelets themselves rather than patients. Supplementation of human stored platelets with exogenous S1P may raise S1P levels, whereas high-density lipoprotein 3 or apolipoprotein A-I, abundantly available in Cohn fraction V from albumin production, could block EV release from platelets.58,59 Selection of platelet products with the shortest storage duration should be considered for recipients at high risk for TRALI. Considering that many of these therapeutic interventions are already approved for clinical use in different diseases, these interventions may prove translatable strategies to prevent TRALI reactions.
Presented in part in abstract form at the annual Experimental Biology Meeting, San Diego, CA, 21-25 April 2018.
For original data, please contact mark.mcvey@utoronto.ca.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Canadian Blood Services (NET-CAD) for providing human platelet products.
This work was supported by a grant from the Canadian Blood Services (CBS) in partnership with the Canadian Institutes of Health Research (CIHR; CIHR-TRA201403-WK-325399) (W.M.K.) and a Health Canada/CBS Priority Grant (2013JS-N-317899) (J.W.S. and W.M.K.). M.J.M. was supported by a CIHR Vanier scholarship.
The flow cytometric expertise of C.S. on EVs allowed and inspired the authors to pursue this work, which is dedicated to his memory.
Authorship
Contribution: M.J.M., C.S., A.T., J.W.S., and W.M.K. designed and interpreted experiments; M.J.M., M.M., S.S., S.W., C.S., M.K., V.S., and A.T.-F. conducted experiments; C.A. contributed vital reagents; and M.J.M. and W.M.K. wrote and edited the paper, which was also edited by J.W.S.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Chris Spring died 3 November 2019.
Correspondence: Wolfgang M. Kuebler, Institute of Physiology, Charité-Universitätsmedizin, Charitéplatz 1, 10117 Berlin, Germany; e-mail: wolfgang.kuebler@charite.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal