

Key Points
TSP-1–deficient platelets show increased bleeding times and diminished thrombosis in vivo.
Platelet-derived TSP-1 corrects hemostasis and is associated with CD36-dependent platelet PGI2 hyposensitivity and diminished cAMP signaling.

Abstract
Thrombospondin-1 (TSP-1) is released by platelets upon activation and can increase platelet activation, but its role in hemostasis in vivo is unclear. We show that TSP-1 is a critical mediator of hemostasis that promotes platelet activation by modulating inhibitory cyclic adenosine monophosphate (cAMP) signaling. Genetic deletion of TSP-1 did not affect platelet activation in vitro, but in vivo models of hemostasis and thrombosis showed that TSP-1–deficient mice had prolonged bleeding, defective thrombosis, and increased sensitivity to the prostacyclin mimetic iloprost. Adoptive transfer of wild-type (WT) but not TSP-1−/− platelets ameliorated the thrombotic phenotype, suggesting a key role for platelet-derived TSP-1. In functional assays, TSP-1–deficient platelets showed an increased sensitivity to cAMP signaling, inhibition of platelet aggregation, and arrest under flow by prostacyclin (PGI2). Plasma swap experiments showed that plasma TSP-1 did not correct PGI2 hypersensitivity in TSP-1−/− platelets. By contrast, incubation of TSP-1−/− platelets with releasates from WT platelets or purified TSP-1, but not releasates from TSP-1−/− platelets, reduced the inhibitory effects of PGI2. Activation of WT platelets resulted in diminished cAMP accumulation and downstream signaling, which was associated with increased activity of the cAMP hydrolyzing enzyme phosphodiesterase 3A (PDE3A). PDE3A activity and cAMP accumulation were unaffected in platelets from TSP-1−/− mice. Platelets deficient in CD36, a TSP-1 receptor, showed increased sensitivity to PGI2/cAMP signaling and diminished PDE3A activity, which was unaffected by platelet-derived or purified TSP-1. This scenario suggests that the release of TSP-1 regulates hemostasis in vivo through modulation of platelet cAMP signaling at sites of vascular injury.
Introduction
The controlled activation of blood platelets at sites of vascular damage is essential for hemostasis. Vascular injury exposes platelets to the prothrombotic extracellular matrix (ECM) proteins von Willebrand factor (vWF), and collagen, which stimulate their transition from a quiescent to an activated state. To moderate excessive activation and return platelets to their quiescent state after transient activation, the endothelium releases prostacyclin (PGI2), which inhibits platelets through a cyclic adenosine monophosphate (cAMP)-dependent signaling cascade.1 This complex signaling system involves enzymes that generate, propagate, and terminate cAMP signaling. PGI2 activates membrane adenylyl cyclases through Gαs-coupled receptors to increase cAMP levels.2 Elevations in cAMP result in the activation of protein kinase A (PKA) isoforms and the subsequent phosphorylation of protein substrates, which underpin the ability of cAMP signaling to control multiple aspects of platelet function.3 Increased platelet cAMP is associated with reductions in Ca2+ mobilization, dense granule secretion, and integrin αIIbβ3 activation and aggregation in vitro,4 as well as reduced platelet accrual at sites of vascular injury in vivo.5
The marginalization of platelets during blood flow facilitates their continual exposure to PGI2 throughout the circulation, ensuring they remain hemostatically inactive. At sites of vascular injury, platelets must overcome the inhibitory effects of PGI2 to ensure rapid hemostasis. This action is achieved through the direct inhibition of cAMP generation by platelet-derived adenosine diphosphate (ADP) binding to Gαi-coupled P2Y12 receptors to inhibit adenylyl cyclase activity.6 Thrombin- and collagen-mediated cAMP hydrolysis may also contribute.7 The reduction in cAMP synthesis in platelets removes the tonic inhibition of cAMP signaling, thereby promoting platelet activation and hemostasis.
TSP-1 is a homotrimeric multidomain glycoprotein present in the ECM, plasma, and platelet α-granules.8-10 The amount of TSP-1 in platelets is estimated to be 0.5 μg/108 platelets, making it one of the most abundant granule proteins.11 Under physiological conditions, TSP-1 plasma levels vary between 50 and 450 ng/mL, but its release from platelets can increase plasma concentrations by 10-fold.10,12,13 TSP-1 binds to platelet glycoprotein IV (CD36) with high affinity,14 but it does not cause platelet activation.15 Rather, it may have a critical role in the regulation of platelet–endothelium crosstalk. Under flow, TSP-1 facilitates platelet–endothelial cell interactions and supports platelet adhesion in vitro in a CD36- and CD47-dependent manner.16 Interestingly, although TSP-1–deficient platelets aggregate normally,17 elegant studies using TSP-1−/− and vWF−/− animals show that TSP-1 contributes to vWF-dependent thrombus formation.18 A second area of TSP-1 biology concerns its ability to influence cyclic nucleotide signaling. Elegant studies by Isenberg et al19 showed that TSP-1 modulated platelet sensitivity to the nitric oxide (NO)–guanosine 3′,5′-cyclic monophosphate (cGMP) inhibitory pathway and increased sensitivity to platelet agonists. Our previous work showed that exogenous TSP-1 could modulate cAMP signaling in vitro.15 Building on these observations, we present evidence that in vivo platelet-derived TSP-1 promotes hemostasis and regulates thrombosis by reducing platelet sensitivity to PGI2.
Materials and methods
Reagents
Experiments with human samples were approved by the Hull York Medical School and the University of Leeds Medical School Research Ethics Boards. All experiments were conducted in accordance with the Declaration of Helsinki.
Experimental animals
CD36−/− mice (from Maria Febbraio, University of Alberta, Edmonton, AB, Canada), TSP-1−/− mice (from The Jackson Laboratory, Bar Harbor, ME), and wild-type (WT) littermates were all on C57BL/6 backgrounds. All procedures were approved by the UK Home Office under the Animals (Scientific Procedures) Act 1986.
Platelet aggregation, flow assays, flow cytometry, intravital microscopy, immunoprecipitation, immunoblotting, phosphodiesterase activity assay, and cAMP measurement
Detailed protocols are described in the supplemental Methods (available on the Blood Web site).
Statistics
Results are expressed as means ± standard error of the mean, unless otherwise stated, and statistical analyses were performed on GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA). Comparisons between groups were performed by using an unpaired, nonparametric Mann-Whitney U test. Statistical significance was accepted at P < .05.
Results
TSP-1–deficient mice display hemostatic defects
We examined the role of platelet-derived TSP-1 in hemostasis and thrombosis using mice deficient in TSP-1 (Figure 1A). In tail bleeding experiments, an indicator of hemostatic capacity, bleeding times for TSP-1−/− mice were significantly increased compared with WT mice (257.3 ± 36 seconds vs 172.5 ± 14 seconds; P < .02) (Figure 1B). Arterial thrombosis in carotid arteries of TSP-1–deficient mice induced by ferric chloride injury was delayed, reduced (mean peak thrombus size at 30 minutes, WT 5586 ± 1407 vs TSP-1−/− 2580 ± 1030 px; P = .01) (Figure 1C), and less stable (supplemental Video 1). Immunoblotting of WT plasma postinjury showed elevated TSP-1 levels in the plasma compared with a noninjured carotid artery (Figure 1D), indicating that platelet activation and thrombosis are associated with TSP-1 secretion. Platelet α-granules are a potential major source of TSP-1 in the vasculature, although leukocytes and endothelial cells may also contribute.20,21

TSP-1 deficiency delays thrombus formation and prolongs bleeding time. (A) Representative immunoblot of TSP-1 from lysates of WT and TSP-1−/− platelets. (B) Animals were counted after cessation of bleeding for 1 minute. Data are presented as scatter plot; every dot represents an individual animal, and the black line indicates mean. N = 7 for WT and N = 8 for TSP-1−/− mice (P < .02). (C) In vivo thrombus formation following ferric chloride injury of the carotid artery of WT mice was compared with that of TSP-1−/− mice (supplemental Videos). Platelets were labeled with Dylight488-conjugated rat anti-CD42b antibody. (i) Representative images of thrombus accrual at indicated times, (ii) representative graph of continuous monitoring of real-time thrombus formation (WT = black line and TSP-1−/− = gray line), and (iii) time to peak thrombus size expressed as mean ± standard deviation of N = 8 WT (black bar) and N = 8 TSP-1−/− (gray bar) mice (*P < .01). (D) Plasma TSP-1 pull-down from noninjured mice and from mice 30 minutes after ferric chloride injury examined by immunoblotting. Representative of 5 mice. (E) Peak thrombus size analysis (20 minutes’ postinjury) after transfusion of WT or TSP-1−/− donor platelets into TSP-1−/− recipient mice followed by ferric chloride injury. Data are presented as mean ± standard deviation and represent N = 5 (*P < .02).
TSP-1 deficiency delays thrombus formation and prolongs bleeding time. (A) Representative immunoblot of TSP-1 from lysates of WT and TSP-1−/− platelets. (B) Animals were counted after cessation of bleeding for 1 minute. Data are presented as scatter plot; every dot represents an individual animal, and the black line indicates mean. N = 7 for WT and N = 8 for TSP-1−/− mice (P < .02). (C) In vivo thrombus formation following ferric chloride injury of the carotid artery of WT mice was compared with that of TSP-1−/− mice (supplemental Videos). Platelets were labeled with Dylight488-conjugated rat anti-CD42b antibody. (i) Representative images of thrombus accrual at indicated times, (ii) representative graph of continuous monitoring of real-time thrombus formation (WT = black line and TSP-1−/− = gray line), and (iii) time to peak thrombus size expressed as mean ± standard deviation of N = 8 WT (black bar) and N = 8 TSP-1−/− (gray bar) mice (*P < .01). (D) Plasma TSP-1 pull-down from noninjured mice and from mice 30 minutes after ferric chloride injury examined by immunoblotting. Representative of 5 mice. (E) Peak thrombus size analysis (20 minutes’ postinjury) after transfusion of WT or TSP-1−/− donor platelets into TSP-1−/− recipient mice followed by ferric chloride injury. Data are presented as mean ± standard deviation and represent N = 5 (*P < .02).
To examine whether platelet-derived TSP-1 was required for thrombosis, we performed an adoptive transfer of WT or TSP-1−/− platelets into TSP-1−/− recipient mice. The infusion of WT platelets led to a partial correction of ferric chloride–induced thrombosis, with peak thrombus size increased (mean peak thrombus size at 30 minutes, 2032 ± 327 vs 3102 ± 342 px; P < .02). In contrast, thrombosis in mice that received an infusion of TSP-1−/− platelets was similar to that observed in TSP-1−/− mice that received no infusion (TSP-1−/− 2032 ± 327 vs TSP-1−/−/TSP-1−/− 1910 ± 220 px; P = .08) (Figure 1E). These data suggest that the absence of platelet-derived TSP-1 accounts for increased bleeding time and diminished thrombosis in TSP-1–deficient mice.
TSP-1 deficiency leads to platelet hypersensitivity to PGI2
Thrombosis involves the rapid accrual of platelets at the site of injury, along with activation of coagulation. In vitro studies showed that platelet aggregation (Figure 2A), integrin αIIbβ3 activation (JonA binding) (Figure 2B), and α-granule secretion (TREM-like transcript 1 expression [TLT-1]) (Figure 2C)22 in response to thrombin or collagen (cross-linked collage-related peptide [CRP-XL] for flow cytometry) was equivalent in both strains of mice. Consistent with the flow cytometry results, we found that the absence of TSP-1 did not affect dense granule secretion (supplemental Figure 1). Analysis of TSP-1–deficient platelets revealed that receptor expression levels of GPIb, GPVI, αIIbβ3, and CD36 (supplemental Figure 2) were comparable to WT platelets. Consistent with previous studies, TSP-1−/− platelet α-granule contents, assessed by immunoblotting of vWF and fibrinogen, were comparable to those in WT mice (supplemental Figure 3).17 Similarly, immunostaining showed that vWF release from adherent WT and TSP-1−/− platelets was similar (supplemental Figure 4). We measured thrombin generation to determine if the observed phenotype was related to defective secondary hemostasis. Tissue factor–induced thrombin generation was similar in WT and TSP-1−/− plasma (arbitary fluorescence units; WT 50 825 ± 15 216 vs TSP-1−/− 59 017 ± 11 874; P = .17) (supplemental Figure 5). Maximal thrombin generation and time to reach maximal generation were also comparable in WT and TSP-1−/− mice (not shown).
![TSP-1 deficiency has no effect on platelet activation. (A) (i) Platelet-rich plasma (PRP) from WT (red bars) and TSP-1−/− (blue bars) mice were stimulated with thrombin (0.01-0.1 U/mL), and platelet aggregation was measured under constant stirring (1000 rpm) at 37°C for 4 minutes. Percentage aggregation is presented as mean ± standard error of the mean (SEM); N = 5). (ii) PRP from WT and TSP-1−/− mice were stimulated with thrombin (0.001-0.025 U/mL) for 20 minutes, and JonA binding was assessed by using flow cytometry. Data are presented as percent positive platelets (N = 5). (iii) as in (ii), except TLT-1 surface expression (α-granule secretion) was measured. Data are presented as percent positive cells (n = 5). (B) as in (A) except (i), where aggregation was induced by collagen (2.5 or 10 μg/mL), and (ii-iii), where CRP-XL 1 and 5 µg/mL were used (N = 4). (C) PRP from WT (red line) and TSP-1−/− (blue line) mice (incubated with apyrase [2 U/mL]) were treated with PGI2 (0-100 nM) for 1 minute before stimulation with collagen (10 μg/mL), and platelet (N = 5) aggregation was recorded over 3 minutes. (i) Percent aggregation is presented as mean ± SEM (P < .05; N = 5). (ii) Representative traces using PGI2 (5 nM). (D) PRP from WT (red) and TSP-1−/− (blue) mice (incubated with apyrase [2 U/mL]) were treated with PGI2 (10 nM) for 1 minute before stimulation with CRP-XL (10 μg/mL) for 20 minutes, and JonA binding was measured. Data are presented as percent positive platelets; mean ± SEM (N = 6; *P < .05, Mann-Whitney U test2). (E) as in (D), except surface expression of TLT-1 was measured by flow cytometry (N = 6; *P < .02).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/5/10.1182_blood.2020005382/1/m_bloodbld2020005382f2.png?Expires=1769139033&Signature=X3AvCKPzsjL3b0GAenbU2zm0Q4QsxZM7LhaGOg~QMoDEXTLNzMq880ppjeKPoPIG1jS2AyJEyd1TccAWfjNpVQD4RKoMfpQvoNlSq-pzd97PbVrRSCBCOwzrUfzkpsxyd-esuF15Elb5OyspMaSlZANFwAcv9sEc5s3-X~SSNHog9fWF71NDzvV0crjYp-QfXRghny3jdhDOfXByIlBn13fc4O89urvgu8278866CtILv9zNxHEah4cI5CTniVTJVkgxp9AQglNrKhSTkoeeVwo2OZe2W-DV-bMalKbyOgDXvtmMAn1KI-Xdd9MHv7e5~Iu8ob5d4akcFLE7z~QjWg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TSP-1 deficiency has no effect on platelet activation. (A) (i) Platelet-rich plasma (PRP) from WT (red bars) and TSP-1−/− (blue bars) mice were stimulated with thrombin (0.01-0.1 U/mL), and platelet aggregation was measured under constant stirring (1000 rpm) at 37°C for 4 minutes. Percentage aggregation is presented as mean ± standard error of the mean (SEM); N = 5). (ii) PRP from WT and TSP-1−/− mice were stimulated with thrombin (0.001-0.025 U/mL) for 20 minutes, and JonA binding was assessed by using flow cytometry. Data are presented as percent positive platelets (N = 5). (iii) as in (ii), except TLT-1 surface expression (α-granule secretion) was measured. Data are presented as percent positive cells (n = 5). (B) as in (A) except (i), where aggregation was induced by collagen (2.5 or 10 μg/mL), and (ii-iii), where CRP-XL 1 and 5 µg/mL were used (N = 4). (C) PRP from WT (red line) and TSP-1−/− (blue line) mice (incubated with apyrase [2 U/mL]) were treated with PGI2 (0-100 nM) for 1 minute before stimulation with collagen (10 μg/mL), and platelet (N = 5) aggregation was recorded over 3 minutes. (i) Percent aggregation is presented as mean ± SEM (P < .05; N = 5). (ii) Representative traces using PGI2 (5 nM). (D) PRP from WT (red) and TSP-1−/− (blue) mice (incubated with apyrase [2 U/mL]) were treated with PGI2 (10 nM) for 1 minute before stimulation with CRP-XL (10 μg/mL) for 20 minutes, and JonA binding was measured. Data are presented as percent positive platelets; mean ± SEM (N = 6; *P < .05, Mann-Whitney U test2). (E) as in (D), except surface expression of TLT-1 was measured by flow cytometry (N = 6; *P < .02).
TSP-1 deficiency has no effect on platelet activation. (A) (i) Platelet-rich plasma (PRP) from WT (red bars) and TSP-1−/− (blue bars) mice were stimulated with thrombin (0.01-0.1 U/mL), and platelet aggregation was measured under constant stirring (1000 rpm) at 37°C for 4 minutes. Percentage aggregation is presented as mean ± standard error of the mean (SEM); N = 5). (ii) PRP from WT and TSP-1−/− mice were stimulated with thrombin (0.001-0.025 U/mL) for 20 minutes, and JonA binding was assessed by using flow cytometry. Data are presented as percent positive platelets (N = 5). (iii) as in (ii), except TLT-1 surface expression (α-granule secretion) was measured. Data are presented as percent positive cells (n = 5). (B) as in (A) except (i), where aggregation was induced by collagen (2.5 or 10 μg/mL), and (ii-iii), where CRP-XL 1 and 5 µg/mL were used (N = 4). (C) PRP from WT (red line) and TSP-1−/− (blue line) mice (incubated with apyrase [2 U/mL]) were treated with PGI2 (0-100 nM) for 1 minute before stimulation with collagen (10 μg/mL), and platelet (N = 5) aggregation was recorded over 3 minutes. (i) Percent aggregation is presented as mean ± SEM (P < .05; N = 5). (ii) Representative traces using PGI2 (5 nM). (D) PRP from WT (red) and TSP-1−/− (blue) mice (incubated with apyrase [2 U/mL]) were treated with PGI2 (10 nM) for 1 minute before stimulation with CRP-XL (10 μg/mL) for 20 minutes, and JonA binding was measured. Data are presented as percent positive platelets; mean ± SEM (N = 6; *P < .05, Mann-Whitney U test2). (E) as in (D), except surface expression of TLT-1 was measured by flow cytometry (N = 6; *P < .02).
We have previously shown that exogenous TSP-1 promotes activation of human platelets by inhibiting cAMP signaling.15 To explore whether this action contributed to our observations in vivo, we evaluated the effect of PGI2 on platelet aggregation in the presence of apyrase to prevent secreted ADP affecting cAMP signaling (supplemental Figure 6).23 TSP-1 secretion from α-granules was not impaired by apyrase (supplemental Figure 7). PGI2 (0-100 nM) caused a concentration-dependent inhibition of collagen-induced aggregation in both WT and TSP-1−/− mice. However, TSP-1−/− platelets showed increased sensitivity to PGI2 with a significantly lower 50% effective concentration (10.4 ± 1 nM vs 28.7 ± 2.4 nM; P < .001) (Figure 2C). Pretreatment of platelet-rich plasma with PGI2 (10 nM) caused a significant reduction in the number of platelets expressing activated αIIbβ3 integrin and TLT-1 in response to CRP-XL in both strains (Figure 2D-E). Inhibition was greater in TSP-1−/− platelets than in WT platelets under the same conditions for integrin activation (WT 62.5 ± 10 vs TSP-1−/− 35.5 ± 8.4; P = .05) and TLT-1 expression (WT 75.5 ± 5 vs TSP-1−/− 50.4 ± 7; P = .018). Similar data were obtained when thrombin was used to stimulate platelet activation (supplemental Figure 8). TSP-1−/− platelets are therefore hypersensitive to PGI2, resulting in altered expression of αIIbβ3 and TLT-1 after activation.
Regulation of PGI2 signaling requires the release of TSP-1 from α-granules
To better understand the relative contribution of platelet-derived and plasma TSP-1 to the platelet response to PGI2, we performed plasma swap experiments. PGI2-induced inhibition of aggregation of WT platelets resuspended in plasma from either WT or TSP-1−/− mice was indistinguishable (Figure 3A). However, inhibition by PGI2 of TSP-1−/− platelets resuspended in TSP-1−/− plasma was greater than WT platelets in WT plasma (61 ± 4% vs 40.3 ± 3.2%; P < .05). To assess the ability of TSP-1−/− platelets to respond to PGI2 in TSP-1–deficient plasma, reciprocal experiments were performed. Inhibition by PGI2 of TSP-1−/− platelets resuspended in TSP-1−/− plasma was indistinguishable from TSP1−/− platelets that were resuspended in WT plasma (40 ± 2.8% vs 38 ± 5.7%).

Platelet-derived, but not plasma-derived, TSP-1 modulates the platelet response to PGI2. (A) WT and TSP-1−/− washed platelets were resuspended in either WT or TSP-1−/− plasma to a concentration of 2 × 108 platelets/mL in the presence of apyrase (2 U/mL). Platelets, in the presence and absence of PGI2 (5 nM), were stimulated with collagen (10 μg/mL), and aggregation was measured under constant stirring (1000 rpm) at 37°C for 4 minutes. (i) Representative traces and (ii) percent aggregation are presented as mean ± standard error of the mean (SEM). (N = 5; **P < .01 compared with platelets treated with collagen and PGI2, Mann-Whitney U test.) (B) Platelet-rich plasma from TSP-1−/− mice (treated with apyrase) were incubated with releasates from WT platelets or TSP-1−/− platelets (or platelet poor plasma 1:10 vol/vol), then stimulated with collagen (10 μg/mL) in the presence or absence of PGI2 (5 nM). (i) Representative traces and (ii) percent aggregation are presented as mean ± SEM (N = 5; *P < .05 compared with platelets treated with collagen and PGI2, Mann-Whitney U test.) (C) as in (B), except platelets were stimulated with collagen (2 μg/mL) in the absence of PGI2. (D) Platelet-rich plasma from TSP-1−/− mice (treated with apyrase) were incubated with human platelet-derived TSP-1 (hTSP-1) before treatment with PGI2 (5 nM) for 1 minute and stimulation with collagen (10 μg/mL). (i) Representative traces; (ii) percent aggregation is presented by mean ± SEM (N = 4, *P < .05, Mann-Whitney U test). Intracellular cAMP levels are presented as mean ± SEM (N = 5, *P < .05, Mann-Whitney U test). NS, not significant.
Platelet-derived, but not plasma-derived, TSP-1 modulates the platelet response to PGI2. (A) WT and TSP-1−/− washed platelets were resuspended in either WT or TSP-1−/− plasma to a concentration of 2 × 108 platelets/mL in the presence of apyrase (2 U/mL). Platelets, in the presence and absence of PGI2 (5 nM), were stimulated with collagen (10 μg/mL), and aggregation was measured under constant stirring (1000 rpm) at 37°C for 4 minutes. (i) Representative traces and (ii) percent aggregation are presented as mean ± standard error of the mean (SEM). (N = 5; **P < .01 compared with platelets treated with collagen and PGI2, Mann-Whitney U test.) (B) Platelet-rich plasma from TSP-1−/− mice (treated with apyrase) were incubated with releasates from WT platelets or TSP-1−/− platelets (or platelet poor plasma 1:10 vol/vol), then stimulated with collagen (10 μg/mL) in the presence or absence of PGI2 (5 nM). (i) Representative traces and (ii) percent aggregation are presented as mean ± SEM (N = 5; *P < .05 compared with platelets treated with collagen and PGI2, Mann-Whitney U test.) (C) as in (B), except platelets were stimulated with collagen (2 μg/mL) in the absence of PGI2. (D) Platelet-rich plasma from TSP-1−/− mice (treated with apyrase) were incubated with human platelet-derived TSP-1 (hTSP-1) before treatment with PGI2 (5 nM) for 1 minute and stimulation with collagen (10 μg/mL). (i) Representative traces; (ii) percent aggregation is presented by mean ± SEM (N = 4, *P < .05, Mann-Whitney U test). Intracellular cAMP levels are presented as mean ± SEM (N = 5, *P < .05, Mann-Whitney U test). NS, not significant.
To substantiate the role of platelet-derived TSP-1, we treated TSP-1−/− platelets with releasates from either WT or TSP-1−/− platelets. WT releasates, containing TSP-1 (supplemental Figure 7), reduced platelet sensitivity to PGI2 as aggregation increased from 17.3 ± 2.6% to 41 ± 4.5% (P < .01) (Figure 3B). In contrast, TSP-1−/− releasates did not affect PGI2-induced platelet inhibition, indicating that the loss of TSP-1 (and not other released factors) was likely responsible for our observations. TSP-1 released from platelets binds rapidly (within 1 minute) to platelet surface receptors,24 but, importantly, treatment of TSP-1−/− platelets with WT releasate neither initiates nor potentiates platelet aggregation in the absence of PGI2 (Figure 3C). When TSP-1−/− platelets, adhered to collagen, were treated with WT releasate, we found that TSP-1 from the releasate could bind to the surface of TSP-1−/− platelets (supplemental Figure 9), confirming that these platelets retain their ability to bind TSP-1. Finally, incubation of TSP-1−/− platelets with purified human TSP-1 also caused a partial reduction of platelet sensitivity to PGI2 (percent aggregation, 11.7 ± 2.2 vs 22.3 ± 1.4; P = .01) (Figure 3D). Thus, our data suggest that platelet-derived TSP-1 has the potential to alter platelet sensitivity to PGI2.
TSP-1 regulates cAMP signaling in platelets
Given the hypersensitivity of TSP-1−/− platelets to PGI2, we assessed the cAMP signaling pathway. Surprisingly, no significant difference was found in cAMP concentrations between WT and TSP-1−/− platelets after treatment with PGI2 (Figure 4A). Importantly, human TSP-1 added exogenously to TSP-1−/− platelets still prevents PGI2-induced cAMP accrual (Figure 4B). Thus, we reasoned that TSP-1 must be released from α-granules to signal in an autocrine and/or paracrine fashion. Hence, platelets were treated with PGI2 (10 nM), then stimulated with collagen (10 μg/mL) in the presence of apyrase for measurement of cAMP. In WT platelets, PGI2-induced cAMP formation was significantly reduced after stimulation with collagen (1850 ± 120 vs 1115 ± 295 cAMP fmol/10 × 7; P < .04), but activation of TSP-1−/− platelets had no effect on cAMP levels (2048 ± 180 vs 2016 ± 101 cAMP fmol/10 × 7; P < .4) (Figure 4C). These data suggest that released TSP-1 may modulate PGI2/cAMP signaling via increased hydrolysis rather than reduced synthesis of cAMP. To verify this theory, we measured the activity of phosphodiesterase 3A (PDE3A), an enzyme responsible for cAMP degradation in platelets.25 The activity of immunoprecipitated PDE3A was not affected by the activation status of platelets (supplemental Figure 10), and it retained sensitivity to milrinone (supplemental Figure 11). Collagen caused a significant increase in PDE3A activity over basal in WT platelets, but this action was muted in TSP-1−/− platelets (Figure 4D). NO-induced increases in cGMP may inhibit platelet PDE3A26 and, under some conditions, PDE3A may hydrolyze cGMP. We therefore measured cGMP to ensure that it was not affected under our experimental conditions. The NO donor S-nitrosoglutathione (10 μM) caused a significant increase in cGMP, and neither TSP-1 nor PGI2 alone had any effect. Moreover, TSP-1 did not inhibit basal cGMP concentrations (supplemental Figure 12), indicating that changes in cGMP were not involved in our observations.

Platelet-derived TSP-1 modulates intracellular cAMP levels by increasing PDE3 activity. (A) WT (red) and TSP-1−/− (blue) platelets (2 × 108 platelets/mL) were treated with PGI2 (0-50 nM) for 1 minute and lysed before intracellular cAMP concentrations were measured. Mean ± standard error of the mean (SEM) (N = 5). (B) TSP-1−/− (red) platelets (2 × 108 platelets/mL) were treated with PGI2 (100 nM) for 1 minute in the presence or absence of human TSP-1 (hTSP-1; 10 μg/mL) and lysed before intracellular cAMP concentrations were measured. Mean ± SEM (N = 5, *P < .05, Mann-Whitney U test2). (C) WT (red bars) and TSP-1−/− (blue bars) platelets (2 × 108 platelets/mL) incubated with apyrase were treated with PGI2 (10 nM) alone for 2 minutes or stimulated with collagen (10 μg/mL) 1 minute after PGI2 treatment. Reactions were stopped with lysis buffer, and intracellular cAMP levels were measured by using enzyme-linked immunosorbent assay. Intracellular cAMP levels are presented as mean ± SEM (N = 6; *P < .05, Mann-Whitney U test2). (D) WT (red bars) and TSP-1−/− (blue bars) platelets (5 × 108 platelets/mL) incubated with apyrase were stimulated with collagen (20 μg/mL) for 1 minute before stopping the reaction with lysis buffer. PDE3A was immunoprecipitated, and enzyme activity was measured. Data are presented as percent activity above basal and given as mean ± SEM (N = 6; P < .05). (E) Whole blood from WT (red bars) and TSP-1−/− (blue bars) mice were incubated with apyrase and treated with PGI2 (10 nM) alone for 2 minutes or stimulated with CRP-XL (10 μg/mL) for 1 minute after PGI2 treatment. Blood was fixed, permeabilized, and incubated with anti-VASPSer157 followed by secondary fluorescent-conjugate (Alexa 647) and analyzed by flow cytometry. Data are presented as mean ± SEM fold increase in phosph-VASPSer157 over basal (N = 4; P < .03, *P < .05, Mann-Whitney U test).
Platelet-derived TSP-1 modulates intracellular cAMP levels by increasing PDE3 activity. (A) WT (red) and TSP-1−/− (blue) platelets (2 × 108 platelets/mL) were treated with PGI2 (0-50 nM) for 1 minute and lysed before intracellular cAMP concentrations were measured. Mean ± standard error of the mean (SEM) (N = 5). (B) TSP-1−/− (red) platelets (2 × 108 platelets/mL) were treated with PGI2 (100 nM) for 1 minute in the presence or absence of human TSP-1 (hTSP-1; 10 μg/mL) and lysed before intracellular cAMP concentrations were measured. Mean ± SEM (N = 5, *P < .05, Mann-Whitney U test2). (C) WT (red bars) and TSP-1−/− (blue bars) platelets (2 × 108 platelets/mL) incubated with apyrase were treated with PGI2 (10 nM) alone for 2 minutes or stimulated with collagen (10 μg/mL) 1 minute after PGI2 treatment. Reactions were stopped with lysis buffer, and intracellular cAMP levels were measured by using enzyme-linked immunosorbent assay. Intracellular cAMP levels are presented as mean ± SEM (N = 6; *P < .05, Mann-Whitney U test2). (D) WT (red bars) and TSP-1−/− (blue bars) platelets (5 × 108 platelets/mL) incubated with apyrase were stimulated with collagen (20 μg/mL) for 1 minute before stopping the reaction with lysis buffer. PDE3A was immunoprecipitated, and enzyme activity was measured. Data are presented as percent activity above basal and given as mean ± SEM (N = 6; P < .05). (E) Whole blood from WT (red bars) and TSP-1−/− (blue bars) mice were incubated with apyrase and treated with PGI2 (10 nM) alone for 2 minutes or stimulated with CRP-XL (10 μg/mL) for 1 minute after PGI2 treatment. Blood was fixed, permeabilized, and incubated with anti-VASPSer157 followed by secondary fluorescent-conjugate (Alexa 647) and analyzed by flow cytometry. Data are presented as mean ± SEM fold increase in phosph-VASPSer157 over basal (N = 4; P < .03, *P < .05, Mann-Whitney U test).
To confirm that modulation of cAMP affects downstream signaling, we measured the phosphorylation of vasodilator-stimulated phosphoprotein (VASP), a target for platelet PKA. Here, we used phosphoflow cytometry to discern subtle changes in phosphorylation in the physiological context of whole blood.27 We found that PGI2 increased phosphoVASPSer157 in WT platelets and that phosphorylation was reduced upon stimulation with CRP-XL (fold change, 5.1 ± 0.3 vs 3.4 ± 0.8; P = .03). In contrast, PGI2-induced phosphoVASPSer157was not affected by stimulation with CRP-XL in TSP-1−/− platelets (fold change, 4.9 ± 0.3 vs 4.9 ± 0.08) (Figure 4E). Critically, immunoblotting showed no differences in key components of the cAMP signaling machinery, including PKARI, PKARII, PKAc, protein kinase G, and PDE3A in TSP-1−/− and WT platelets (supplemental Figure 13).
TSP-1 modulation of cAMP signaling requires CD36
We have previously shown that crosstalk exists between cAMP and CD36 signaling pathways.15 To confirm a role for CD36 in transducing the effects of TSP-1, CD36-deficient platelets were used. We reasoned that PGI2 hypersensitivity would not be observed in the absence of this key receptor. Platelet aggregation, TLT-1 expression, and αIIbβ3 activation (JonA binding) in response to collagen/CRP-XL (Figure 5A-C) or thrombin (supplemental Figure 14) in WT and CD36−/− mice were indistinguishable. In contrast, PGI2-mediated inhibition of platelet aggregation was greater in CD36−/− platelets than in WT platelets. Moreover, PGI2-mediated reductions of CRP-XL–mediated TLT-1 surface expression and integrin αIIbβ3 activation were greater in CD36−/− than in WT. Similarly, pretreatment with PGI2 significantly inhibited thrombin-mediated TLT-1 expression and integrin activation.
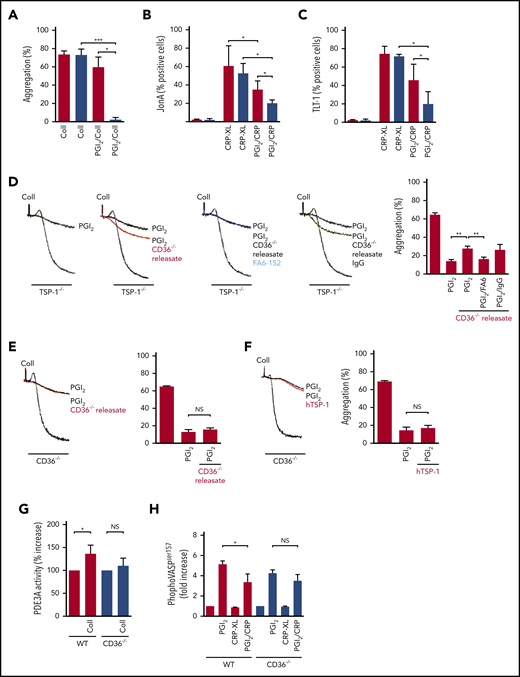
Platelet-derived TSP-1 modulates PDE3 activity in a CD36-dependent manner. (A) Platelet-rich plasma (PRP) from WT and CD36−/− mice (treated with apyrase) were stimulated with collagen (10 μg/mL), in the presence or absence of PGI2 (5 nM), and aggregation was measured. Mean ± standard error of the mean (SEM) (N = 5, **P < .01, Mann-Whitney U test). (B) PRP from WT and CD36−/− mice (treated with apyrase) were treated with PGI2 (10 nM) for 1 minute before stimulation with CRP-XL (5 μg/mL) for 20 minutes, and JonA binding was measured by using flow cytometry (N = 7; P < .05). (C) As in (B), except TLT-1 surface expression was measured (N = 7; P < .05). (D) PRP from TSP-1−/− (treated with apyrase) was stimulated with collagen (10 μg/mL) in the presence and absence of PGI2 (5 nM) for 1 minute and aggregation measured. In some cases, platelets were pretreated with CD36-derived releasates and the CD36 receptor blocking antibody FA6.152 (2 μg/mL) or CD36-derived releasates and immunoglobulin G (IgG) (2 μg/mL). Percentage aggregation is presented as mean ± SEM (N = 5; **P < .01 compared with platelets treated with thrombin and PGI2, Mann-Whitney U test). (E) PRP from CD36−/− mice (treated with apyrase) was stimulated with collagen (10 μg/mL) or treated with PGI2 (5 nM) for 1 minute before stimulation in the presence or absence of CD36-derived releasates. Platelet aggregation is presented as mean ± SEM (N = 6). (F) As in (E), except platelets were treated with human TSP-1 (hTSP-1) (10 µg/mL). (G) WT and CD36−/− platelets (5 × 108 platelets/mL; incubated with apyrase) were stimulated with collagen (10 μg/mL) for 1 minute before stopping the reaction with lysis buffer. PDE3A was immunoprecipitated, and its activity was measured. Data are presented as percent activity above basal activity and presented as mean ± SEM (N = 3; P < .05). (H) Whole blood from WT and CD36−/− mice (incubated with apyrase) was treated with PGI2 (50 nM) alone, CRP-XL (10 μg/mL) alone, or PGI2 followed by CRP-XL. Blood was fixed, permeabilized, and stained for VASP (Ser157) phosphorylation. Quantification is presented as fold increase in median fluorescence intensity over basal (N = 4; *P < .05, Mann-Whitney U test). NS, not significant.
Platelet-derived TSP-1 modulates PDE3 activity in a CD36-dependent manner. (A) Platelet-rich plasma (PRP) from WT and CD36−/− mice (treated with apyrase) were stimulated with collagen (10 μg/mL), in the presence or absence of PGI2 (5 nM), and aggregation was measured. Mean ± standard error of the mean (SEM) (N = 5, **P < .01, Mann-Whitney U test). (B) PRP from WT and CD36−/− mice (treated with apyrase) were treated with PGI2 (10 nM) for 1 minute before stimulation with CRP-XL (5 μg/mL) for 20 minutes, and JonA binding was measured by using flow cytometry (N = 7; P < .05). (C) As in (B), except TLT-1 surface expression was measured (N = 7; P < .05). (D) PRP from TSP-1−/− (treated with apyrase) was stimulated with collagen (10 μg/mL) in the presence and absence of PGI2 (5 nM) for 1 minute and aggregation measured. In some cases, platelets were pretreated with CD36-derived releasates and the CD36 receptor blocking antibody FA6.152 (2 μg/mL) or CD36-derived releasates and immunoglobulin G (IgG) (2 μg/mL). Percentage aggregation is presented as mean ± SEM (N = 5; **P < .01 compared with platelets treated with thrombin and PGI2, Mann-Whitney U test). (E) PRP from CD36−/− mice (treated with apyrase) was stimulated with collagen (10 μg/mL) or treated with PGI2 (5 nM) for 1 minute before stimulation in the presence or absence of CD36-derived releasates. Platelet aggregation is presented as mean ± SEM (N = 6). (F) As in (E), except platelets were treated with human TSP-1 (hTSP-1) (10 µg/mL). (G) WT and CD36−/− platelets (5 × 108 platelets/mL; incubated with apyrase) were stimulated with collagen (10 μg/mL) for 1 minute before stopping the reaction with lysis buffer. PDE3A was immunoprecipitated, and its activity was measured. Data are presented as percent activity above basal activity and presented as mean ± SEM (N = 3; P < .05). (H) Whole blood from WT and CD36−/− mice (incubated with apyrase) was treated with PGI2 (50 nM) alone, CRP-XL (10 μg/mL) alone, or PGI2 followed by CRP-XL. Blood was fixed, permeabilized, and stained for VASP (Ser157) phosphorylation. Quantification is presented as fold increase in median fluorescence intensity over basal (N = 4; *P < .05, Mann-Whitney U test). NS, not significant.
To show that observations with CD36−/− platelets were not due to lack of TSP-1, we showed that releasates from CD36−/− platelets, containing TSP-1 (supplemental Figure 15), decrease the sensitivity of TSP-1–deficient platelets to PGI2 (14 ± 1 vs 26.7 ± 2; P < .0004) (Figure 5D). The effects of releasates were blocked when TSP-1−/− platelets were treated with the CD36-blocking antibody FA6.15228 but not immunoglobulin G (IgG) control. In contrast, CD36−/− platelet releasates did not affect PGI2-induced inhibition of aggregation in CD36−/− mice (Figure 5E). Similar data were obtained when purified human TSP-1 was substituted for the releasate (Figure 5F).
We next examined the cAMP-signaling pathway in CD36−/− mice. Collagen significantly increased PDE3A activity in WT platelets but not in CD36−/− platelets (Figure 5G). Incubation of whole blood from WT mice with PGI2 increased, which was reduced by stimulation of the blood with CRP-XL. Phosphoflow showed that VASP (Ser157) phosphorylation was elevated in CD36−/− platelets in response to PGI2 (Figure 5H), but as with TSP-1−/− platelets, CRP-XL did not affect PGI2-induced phosphorylation. Together, these data suggest that the modulation of cAMP signaling by TSP-1 requires at least partial involvement of surface CD36.
TSP-1 modulates PGI2 control of thrombosis in vitro and hemostasis in vivo
We next examined if altered sensitivity to PGI2 was evident in the blood of TSP-1−/− mice. Here, whole blood from WT or TSP-1−/− mice was treated with apyrase and perfused over immobilized collagen in the presence or absence of PGI2. Consistent with previous studies, we found no difference in thrombus size between TSP-1−/− and WT platelets.16 However, pretreatment with PGI2 caused a greater reduction in thrombus size in TSP-1−/− mice versus control mice (WT 25.1 ± 5.8 vs TSP-1−/− 52.6 ± 05.6%; P = .01) (Figure 6A). Our in vivo studies suggested that thrombi in TSP-1−/− mice were less stable (supplemental Video 2). To test thrombus stability,29 we flowed modified Tyrode’s buffer over preformed thrombi. In both strains of mice, thrombi were relatively stable when challenged with buffer alone. However, in PGI2-supplemented buffer, preformed thrombi in TSP-1−/− blood disintegrated within 4 minutes while WT thrombi remained stable (WT 15 ± 4% vs TSP-1−/− 42 ± 12.9%; P = .05) (Figure 6B). These data highlight the potential importance of TSP-1 in generating and stabilizing thrombi when exposed to PGI2 at high shear flow.
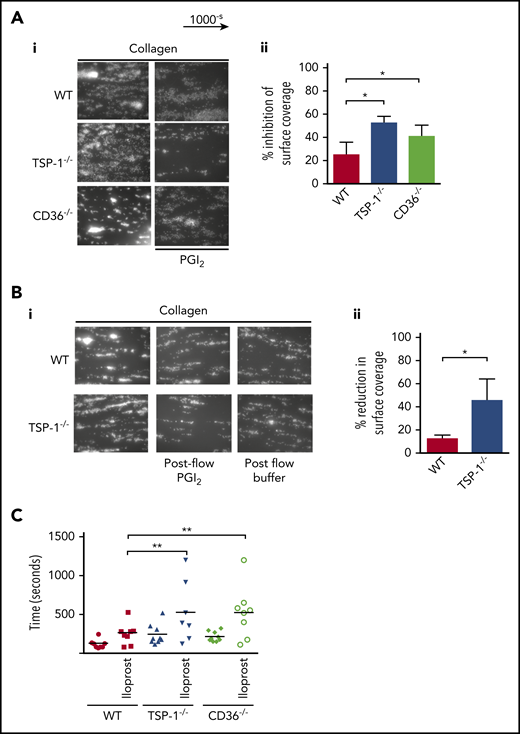
TSP-1−/− and CD36−/− display prolonged bleeding times after iloprost injection. (A) Whole blood was incubated alone or with PGI2 (50 nM) for 2 minutes, then perfused at arterial shear 1000 s−1 for 2 minutes over a collagen matrix (50 µg/mL). Images of adherent platelets were taken by fluorescence microscopy. (i) Representative images of arterial flow experiments, (ii) data presented as inhibition of surface coverage (%), mean ± standard error of the mean (N = 5; *P < .05, Mann-Whitney U test). (B) Whole blood from WT and TSP-1−/− mice was labeled with DIOC6 and perfused over collagen (50 mg/mL) at a shear rate of 1000-s for 2 minutes. The thrombi were subjected to a post-flow with Tyrode’s bovine serum albumin buffer with or without PGI2 (50 nM) at 1000-s for 4 minutes. (i) Representative images at the end of the postflow. (ii) Data are presented as reduction in surface coverage (%); N = 5 (P < .05). (C) WT, TSP-1−/−, and CD36−/− mice were transfused with iloprost (80 ng/mL) for 2 minutes or vehicle control, and bleeding time was examined by using the tail bleeding assay. Animals were counted after complete cessation of bleeding for 1 minute. Data are presented as scatter plot; every dot represents an individual animal, and the black line indicates mean (N = 9; **P < .01).
TSP-1−/− and CD36−/− display prolonged bleeding times after iloprost injection. (A) Whole blood was incubated alone or with PGI2 (50 nM) for 2 minutes, then perfused at arterial shear 1000 s−1 for 2 minutes over a collagen matrix (50 µg/mL). Images of adherent platelets were taken by fluorescence microscopy. (i) Representative images of arterial flow experiments, (ii) data presented as inhibition of surface coverage (%), mean ± standard error of the mean (N = 5; *P < .05, Mann-Whitney U test). (B) Whole blood from WT and TSP-1−/− mice was labeled with DIOC6 and perfused over collagen (50 mg/mL) at a shear rate of 1000-s for 2 minutes. The thrombi were subjected to a post-flow with Tyrode’s bovine serum albumin buffer with or without PGI2 (50 nM) at 1000-s for 4 minutes. (i) Representative images at the end of the postflow. (ii) Data are presented as reduction in surface coverage (%); N = 5 (P < .05). (C) WT, TSP-1−/−, and CD36−/− mice were transfused with iloprost (80 ng/mL) for 2 minutes or vehicle control, and bleeding time was examined by using the tail bleeding assay. Animals were counted after complete cessation of bleeding for 1 minute. Data are presented as scatter plot; every dot represents an individual animal, and the black line indicates mean (N = 9; **P < .01).
Having established that the absence of TSP-1 or CD36 renders platelets hypersensitive to PGI2 in vitro, we confirmed these findings by partially recreating the hemostatic response in vivo with intravenous injection of a PGI2 analogue.30 Iloprost (800 ng/mL) caused a modest but significant increase in the bleeding time of WT mice (120 ± 3 seconds vs 264 ± 62 seconds; P < .03) (Figure 6C). In contrast, it significantly prolonged bleeding times in TSP-1−/− mice (190 ± 15 seconds vs 555 ± 160 seconds; P < .04), whereas the absence of CD36 markedly prolonged bleeding times in CD36−/− mice (214 ± 21 seconds vs 524 ± 118 seconds; P < .01). Phosphoflow performed after iloprost injection revealed an increase in intraplatelet VASP (Ser157) phosphorylation (supplemental Figure 16).
Discussion
The dynamic activation of blood platelets during response to injury requires a rapid suppression of the tonic inhibitory actions of endothelial-derived PGI2 and NO. The current study provides evidence that platelet-derived TSP-1 plays a key role in promoting hemostasis and thrombosis by desensitizing platelets to inhibitory cAMP signaling. Our data show that: (1) TSP-1-deficient mice have a bleeding phenotype, defective thrombosis, and increased sensitivity to the PGI2 mimetic iloprost; (2) transfusion of WT platelets into TSP-1−/− mice corrects defective thrombus formation and improves clot stability; and (3) platelet-derived TSP-1, through CD36, reduces platelet cAMP concentration by activating PDE3A.
Bleeding times in TSP-1−/− mice were significantly increased, suggesting a hemostatic defect. Interestingly, some TSP-1−/− mice showed normal bleeding times, and although the reasons for this outcome are unclear, it is likely linked to variations in thrombus stability we observed both in vitro and in vivo, which can be associated with rebleeding.31 Given that TSP-1 is expressed by monocytes, endothelium, and stromal fibroblasts and that it circulates in the plasma, we explored the possibility that these individual sources play distinct roles in hemostasis,20 focusing on the importance of platelet-derived TSP-1. Although we found that thrombosis was defective in the absence of TSP-1, the use of global rather than tissue-specific TSP-1 knockout mice limited our ability to definitively conclude that platelet-derived TSP-1 is critical. Thus, our observations that thrombotic defects could be ameliorated by the adoptive transfer of WT but not TSP-1−/− platelets, and that TSP-1−/− mice possess normal concentrations of other key adhesive proteins such as vWF and fibrinogen,17 strongly suggest that platelet-derived TSP-1 is largely responsible for thrombus formation and stability in vivo. Given that our data, consistent with previous studies, showed TSP-1−/− platelets to aggregate normally, we were initially surprised by these in vivo findings17,19 because they suggest that the hemostatic defect may be unrelated to platelet activation. We first confirmed that TSP-1 deficiency did not affect coagulation in TSP-1−/− mice, which is particularly relevant given that plasma TSP-1 correlates with thrombin generation32 and potentially increases plasmin generation.33 Having shown that coagulation was not defective, we focused our attention on the mechanisms of platelet activation in vivo. Elegant studies by Sim et al5 showed that modulation of platelet cAMP delayed thrombus formation in vivo. This observation, coupled with our previous work showing that exogenous TSP-1 can reduce platelet sensitivity to PGI2 in vitro,15 led us to examine the in vivo relevance of this pathway. To approach this issue, we used the PGI2 mimetic iloprost, which is known to modulate platelet function in vivo.34 As expected, we found that iloprost increased bleeding times in both strains but that absence of TSP-1 caused significantly longer bleeding times, suggesting that these animals are hypersensitive to platelet cAMP. Interestingly, our in vitro experiments revealed that the phenotype was related to thrombus stability. In agreement with our recent report,35 flow experiments show that PGI2 can induce the dissolution of preformed thrombi but that this is more acute in the absence of TSP-1. The significance of this finding is not yet clear, but it could reveal a new role for cAMP signaling in controlling thrombosis and will require detailed investigation in the future.
The biology of TSP-1 is extremely complex given that it reportedly interacts with ECM proteins, cell receptors, growth factors, cytokines, and proteases, often simultaneously.36 It is therefore likely that TSP-1 has multiple roles in the hemostatic process, with different pools of TSP-1 having distinct functional roles. Our plasma swap experiments suggest that basal plasma concentrations of TSP-1 do not make a major contribution to platelet regulation by cAMP. This does not preclude plasma TSP-1 regulating other elements of hemostasis, including platelet adhesion and thrombosis.16,18,37 One key factor is that TSP-1 binds promiscuously to plasma proteins, which could either reduce its bioavailability for platelets or, more likely, induce changes in TSP-1 that direct it to specific functions. A recent study showed that neutrophil-mediated cleavage of TSP-1 promotes platelet adhesion and string formation.38 Because TSP-1−/− platelets exhibited increased sensitivity to PGI2 in aggregation assays, which were partly reduced by addition of purified human TSP-1 or releasates from WT platelets (containing TSP-1) but not TSP-1−/− platelets, it suggests that platelet TSP-1 could be directed to this specific function. Clearly, data from our cAMP experiments show that platelet activation, and seemingly TSP-1 secretion, is required to dampen cAMP signaling. Given that TSP-1 diminishes cAMP accumulation upon exposure to PGI2 and this action is blocked in vitro by the PDE3A inhibitor milrinone,15 it is likely that TSP-1 induces the breakdown rather than accumulation of cAMP. To this end, we show that TSP-1 signaling is linked directly to activation of PDE activity in platelets. The physiological relevance of these biochemical assays is strengthened by novel whole-blood signaling experiments, showing clearly that PGI2-cAMP signaling responses, as evidenced by VASP phosphorylation, are maintained in TSP-1−/− mice. These data suggest that released TSP-1 increases PDE3A activity, which accelerates cAMP breakdown, leading to diminished PKA activity and reduced sensitivity to PGI2. In the absence of TSP-1, platelets are hypersensitive to inhibitory cAMP signaling. We observed that sensitivity to PGI2 never fully recovers after the addition of either recombinant or platelet-derived TSP-1, for reasons that are unclear. Because we omitted ADP signaling from our experiments, we anticipate that a better recovery could be achieved. It is also possible that in the complex conditions found in vivo, platelet-derived TSP-1 would require processing to be fully effective,36 or that it engages other partner proteins which affect cAMP availability such as multidrug resistance proteins (MRP4) that promote cAMP efflux from platelets39 ; however, these possibilities require further exploration.
Several structurally and functionally distinct receptors for TSP-1 have been identified on platelets, including integrins αvβ3 and αIIbβ3, CD36, and integrin-associated protein (IAP or CD47).14,40-42 We have shown that ligation of CD36 by TSP-1 in vitro can modulate platelet sensitivity to cAMP signaling.15,19,43,44 CD36 deficiency (known as the Nakaa– phenotype) is highly prevalent in Asian populations (∼3%) but less common in the western world.45-47 The lack of evidence for a hemostatic defect in CD36-deficient individuals suggests that binding of collagen to other receptors (eg, GPVI) might be sufficient to avoid an overt bleeding diathesis. In this study, we present 3 pieces of evidence to show a link between CD36 and platelet-derived TSP-1 modulation of platelet function. First, CD36-deficient platelets, as with platelets deficient in TSP-1−/−, display a markedly increased sensitivity to PGI2, which interestingly cannot be rectified by platelet releasates or purified TSP-1. Second, reduced cAMP signaling and increased PDE3A activity in response to platelet activation in the platelets of WT animals is absent in CD36−/− platelets. Finally, the ability of platelet releasates, containing TSP-1, to increase platelet sensitivity to cAMP was blocked by a CD36-blocking antibody. Gosh et al48 showed that, as with TSP-1–deficient mice, CD36−/− mice display delayed thrombus formation in ferric chloride–injured arteries. The authors concluded that CD36 ligands are generated after vascular injury and that signals from these ligands contribute to thrombus formation and stability. Our results show that TSP-1, which is generated during vascular damage, is a likely ligand and that CD36 is required, at least in part, for TSP-1 mediated “disinhibition” of PGI2 signaling; however, given the multivalent nature of TSP-1, it is likely that other receptors may contribute to the modulation of platelet function. The mechanisms underpinning these effects likely involve CD36-mediated tyrosine kinase pathways that are activated by TSP-1 and other ligands.7,15,44,49,50
Our data suggest that in circulating platelets, TSP-1 is localized in α-granules, limiting its availability. At sites of vascular injury, activated platelets release TSP-1, which acts to suppress inhibitory cAMP signaling and promote hemostasis. Our data show that TSP-1 can alter this balance to promote platelet activation, falling in line with previous studies showing that TSP-1−/− platelets exhibit higher sensitivity to inhibition by NO.19 We postulate that control of cAMP and cGMP signaling by TSP-1 represents an important mechanism-effective hemostasis. However, this mechanism is potentially subverted by circulating pathological ligands such as oxidized low-density lipoprotein to promote thrombosis.43,44 The clinical importance of the observations with TSP-1 must now be elucidated, given the increasing interest in the development of therapeutic strategies targeting TSP-1 in advanced primary cancers51 and the prothrombotic platelets generated by these therapies.52 Our data and that of others show that TSP-1 has multifaceted roles in thrombosis and hemostasis, which should be considered in detail when developing new drug interventions.
Requests for data sharing should be e-mailed to the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This project was funded by the British Heart Foundation (PG/13/90/20578, PG/12/49/29441, FS/18/75/33978, and FS/19/10/34128) and the Rotations Program of the Medical Faculty of RWTH Aachen University.
Authorship
Contribution: A.A. designed and performed experiments, analyzed data, and wrote the manuscript; M.B, K.S.W., B.E.J.S., and B.A.W., performed experiments; M.F. provided essential materials; A.W.P. designed the research; and K.M.N. designed the research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Khalid M. Naseem, Leeds Institute of Cardiovascular & Metabolic Medicine, The LIGHT Laboratories, Clarendon Way, University of Leeds, Leeds LS2 9N, United Kingdom; e-mail: k.naseem@leeds.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal