

TO THE EDITOR:
Vacuolization of myeloid and erythroid precursors in the bone marrow (BM) has been recently identified as a hallmark feature of a new adult-onset inflammatory syndrome called VEXAS (vacuoles, E1 enzyme, X linked, autoinflammatory, somatic). This syndrome is genetically associated with somatic mutations affecting methionine 41 of the X-linked gene UBA1.1 The morphologic finding of vacuoles in the context of an underlying molecular lesion in VEXAS raises the question of the presence of vacuolization of hematopoietic precursors (HPs) in other clinical conditions. Although this morphologic feature is well established in lymphoid malignancies such as Burkitt lymphoma/leukemia, it is not frequent in myeloid pathologies. Notably, it has been occasionally encountered in patients with myeloid neoplasms (MNs) in association with karyotypic abnormalities and general higher-risk features as well as in patients with other benign conditions such as alcoholism, protein-losing enteropathy, and typical hypocupremia.2-6 Therefore, vacuolization of HPs seems to be typical but not pathognomonic in VEXAS.
Through chart review of patients enrolled at The Cleveland Clinic Foundation between 2005 and 2020, we identified 24 cases with overt presence of vacuoles in HPs among 11 772 BM specimens. All patients underwent BM evaluation as part of the initial workup and/or follow-up for an MN or to define an unexplained cytopenia (supplemental Figure 1; supplemental Appendix, available on the Blood Web site). Ethical approval was obtained from the institutional review board at The Cleveland Clinic Foundation, along with patients’ written consent to participate to the study. All procedures were carried out in accordance with guidelines set forth by the Declaration of Helsinki.
A majority of patients were male (17 [70%] of 24), with a median age of 65 years (range, 9-92 years; Table 1). Overall, more than half of the patients (14 of 24) showed vacuoles in erythroid (n = 7) or myeloid (n = 2) precursors or both (n = 5), whereas in 10 patients, vacuolization was restricted to myeloid blasts (blast-only). The vacuoles appeared during the course of disease (MDS or AML) rather than at initial specimen evaluation in 15 patients; vacuoles in 9 of these patients showed a blast-only pattern (vs only 1 patient whose BM was collected at the onset of disease; 60% vs 11%; P = .03; Figure 1A). Of note, these 15 MNs were characterized by higher-risk features (80% of cases had at least 1 cytogenetic abnormality vs 33% in the 9 remaining cases collected at onset; P = .04), history of cancer treatment, or antecedent diagnosis of a myeloproliferative neoplasm and did not carry a UBA1 mutation.
Patient characteristics
| UPN | Sex | Age, y | Type of vacuoles | Timing | Diagnosis | WHO classification* | Cytogenetics | Genetic alterations (VAF %)† | Associated conditions |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 65 | MYE/E | Onset | MDS | MDS-MLD | NK | DNMT3A (26), UBA1‡ | Sweet syndrome, VT, VEXAS |
| 2 | M | 62 | MYE/E | Onset | MDS | MDS-MLD | NK | DNMT3A (4) | RA |
| 3 | F | 76 | MYE | Onset | MDS | MDS-EB-2 | CK | NA | Ulcerative colitis |
| 4 | F | 67 | B | Follow-up | tr-MN | tr-MN | CK | TP53 (34) | Graves’ disease |
| 5 | M | 76 | B | Follow-up | sAML | AML-MRC | CK | ASXL1 (52), U2AF1 (9) | |
| 6 | M | 66 | MYE/E | Onset | MDS | MDS-SLD | NK | UBA1‡ | Sweet syndrome, RA, VT, VEXAS |
| 7 | M | 86 | B | Follow-up | tr-MN | tr-MN | CK | ASXL1 (36), IDH2 (19), SF3B1 (21), TET2 (4), TP53 (23) | Previous diagnosis of CLL |
| 8 | M | 64 | E | Follow-up | sAML | AML-MRC | CK | WT | Previous diagnosis of MDS |
| 9 | M | 64 | B | Follow-up | sAML | AML-MRC | CK | JAK2 (36) | Previous diagnosis of ET |
| 10 | M | 87 | B | Follow-up | MDS | MDS-EB-2 | NK§ | 21q21.1-q22.3 LOH§ | |
| 11 | F | 73 | MYE/E | Onset | CD | NA | NK | NA | Alcohol abuse, hepatopathy |
| 12 | M | 65 | E | Follow-up | MDS | MDS-EB-2 | CK | WT | |
| 13 | M | 9 | E | Onset | CS | NA | NK | NA | Complex congenital syndrome |
| 14 | F | 58 | B | Follow-up | sAML | AML-MRC | CK | CUX1 (73) | Previous diagnosis of MDS |
| 15 | M | 42 | MYE | Onset | CD | NA | NK | NA | HIV, celiac disease |
| 16 | M | 92 | E | Onset | sAML | AML-MRC | CK | NA | Previous diagnosis of MDS, HT |
| 17 | F | 76 | B | Onset | MDS | MDS-EB-2 | CK | NA | Vasculitis |
| 18 | F | 49 | E | Follow-up | MDS | MDS-RS-MLD | NK | BCOR (46) | |
| 19 | M | 65 | B | Follow-up | sAML | AML-MRC | +8 | IDH1 (43), RUNX1 (39) | Previous diagnosis of MDS |
| 20 | M | 63 | B | Follow-up | tr-MN | tr-MN | CK | NA | Previous diagnosis of FL |
| 21 | F | 75 | E | Follow-up | tr-MN | tr-MN | CK | DNMT3A (34) | Previous diagnosis of FL |
| 22 | M | 65 | MYE/E | Follow-up | MDS | MDS-SLD | NK | DDX41 (45), DNMT3A (14), PHF6 (15) | |
| 23 | M | 65 | B | Follow-up | sAML | AML-MRC | CK | DNMT3A (44), JAK2 (8), TET2 (46), TET2 (43) | Previous diagnosis of MDS |
| 24 | M | 85 | E | Follow-up | MDS | MDS-MLD | +11 | NA |
| UPN | Sex | Age, y | Type of vacuoles | Timing | Diagnosis | WHO classification* | Cytogenetics | Genetic alterations (VAF %)† | Associated conditions |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 65 | MYE/E | Onset | MDS | MDS-MLD | NK | DNMT3A (26), UBA1‡ | Sweet syndrome, VT, VEXAS |
| 2 | M | 62 | MYE/E | Onset | MDS | MDS-MLD | NK | DNMT3A (4) | RA |
| 3 | F | 76 | MYE | Onset | MDS | MDS-EB-2 | CK | NA | Ulcerative colitis |
| 4 | F | 67 | B | Follow-up | tr-MN | tr-MN | CK | TP53 (34) | Graves’ disease |
| 5 | M | 76 | B | Follow-up | sAML | AML-MRC | CK | ASXL1 (52), U2AF1 (9) | |
| 6 | M | 66 | MYE/E | Onset | MDS | MDS-SLD | NK | UBA1‡ | Sweet syndrome, RA, VT, VEXAS |
| 7 | M | 86 | B | Follow-up | tr-MN | tr-MN | CK | ASXL1 (36), IDH2 (19), SF3B1 (21), TET2 (4), TP53 (23) | Previous diagnosis of CLL |
| 8 | M | 64 | E | Follow-up | sAML | AML-MRC | CK | WT | Previous diagnosis of MDS |
| 9 | M | 64 | B | Follow-up | sAML | AML-MRC | CK | JAK2 (36) | Previous diagnosis of ET |
| 10 | M | 87 | B | Follow-up | MDS | MDS-EB-2 | NK§ | 21q21.1-q22.3 LOH§ | |
| 11 | F | 73 | MYE/E | Onset | CD | NA | NK | NA | Alcohol abuse, hepatopathy |
| 12 | M | 65 | E | Follow-up | MDS | MDS-EB-2 | CK | WT | |
| 13 | M | 9 | E | Onset | CS | NA | NK | NA | Complex congenital syndrome |
| 14 | F | 58 | B | Follow-up | sAML | AML-MRC | CK | CUX1 (73) | Previous diagnosis of MDS |
| 15 | M | 42 | MYE | Onset | CD | NA | NK | NA | HIV, celiac disease |
| 16 | M | 92 | E | Onset | sAML | AML-MRC | CK | NA | Previous diagnosis of MDS, HT |
| 17 | F | 76 | B | Onset | MDS | MDS-EB-2 | CK | NA | Vasculitis |
| 18 | F | 49 | E | Follow-up | MDS | MDS-RS-MLD | NK | BCOR (46) | |
| 19 | M | 65 | B | Follow-up | sAML | AML-MRC | +8 | IDH1 (43), RUNX1 (39) | Previous diagnosis of MDS |
| 20 | M | 63 | B | Follow-up | tr-MN | tr-MN | CK | NA | Previous diagnosis of FL |
| 21 | F | 75 | E | Follow-up | tr-MN | tr-MN | CK | DNMT3A (34) | Previous diagnosis of FL |
| 22 | M | 65 | MYE/E | Follow-up | MDS | MDS-SLD | NK | DDX41 (45), DNMT3A (14), PHF6 (15) | |
| 23 | M | 65 | B | Follow-up | sAML | AML-MRC | CK | DNMT3A (44), JAK2 (8), TET2 (46), TET2 (43) | Previous diagnosis of MDS |
| 24 | M | 85 | E | Follow-up | MDS | MDS-MLD | +11 | NA |
AML, acute myeloid leukemia; B, blasts; CD, copper deficiency; CK, complex karyotype; CLL, chronic lymphocytic leukemia; CS, congenital syndrome; E, erythroid; EB, excess blasts; ET, essential thrombocythemia; F, female; FL, follicular lymphoma; HT, Hashimoto’s thyroiditis; LOH, loss of heterozygosity; M, male; MDS, myelodysplastic syndrome; MLD, multilineage dysplasia; MRC, myelodysplasia-related changes; MYE, myeloid; NA, not applicable/available; NK, normal karyotype; RA, rheumatoid arthritis; RS, ringed sideroblasts; s, secondary; SLD, single-lineage dysplasia; tr, therapy related; VAF, variant allele frequency; UPN, unique patient number; VT, venous thromboembolism; WHO, World Health Organization; WT, wild type.
WHO classification follows the 2016 revision of the WHO classification of MNs and acute leukemia.12
Somatic variants reported are those detected at the time of collection of BM specimens.
UBA1 mutations were detected by Sanger sequencing.
This patient had a copy-neutral LOH involving most of the long arm of chromosome 21 (21q21.1-q22.3) in a small but significant percentage of the cell population. This region would encompass the RUNX1 gene, and the lesion was classified as likely pathogenic.
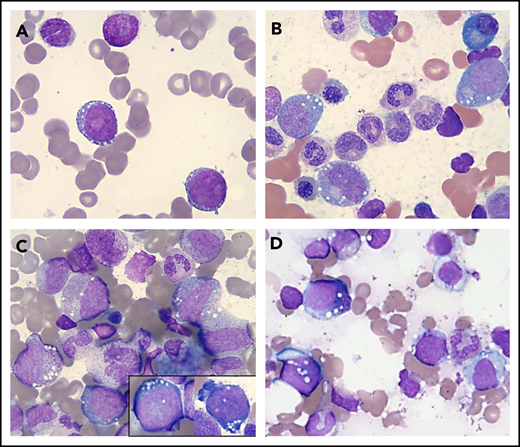
Vacuolization of HPs in various conditions. Wright-Giemsa stains (magnification ×1000) showing a representative example of a "blast-only” pattern of vacuolization in a case with high-risk MDS (vacuoles in 2 blasts) (A) and vacuolization of immature erythroid and myeloid cells in 2 patients with a UBA1 mutation and VEXAS syndrome (B-C) and in a patient with copper deficiency (D).
Vacuolization of HPs in various conditions. Wright-Giemsa stains (magnification ×1000) showing a representative example of a "blast-only” pattern of vacuolization in a case with high-risk MDS (vacuoles in 2 blasts) (A) and vacuolization of immature erythroid and myeloid cells in 2 patients with a UBA1 mutation and VEXAS syndrome (B-C) and in a patient with copper deficiency (D).
We then focused on the cases presenting with vacuolization of myeloid and erythroid precursors at onset (n = 9); taking into consideration demographics and manifestations of the VEXAS syndrome, we identified 6 of 9 cases eligible for hotspot UBA1 mutational screening. Of these, 2 patients (aged 65 and 66 years) tested positive for the UBA1 c.122 T>C p.Met41 mutation (Figure 1B-C; supplemental Figure 2) and showed the typical clinical phenotype, with skin involvement, history of venous thrombosis, and joint pain. Of note, both patients fulfilled the criteria for Sweet syndrome and registered increased levels of C-reactive protein and/or erythrocyte sedimentation rate at onset, with 1 case having a previous diagnosis of rheumatoid arthritis. Moreover, no copper or zinc alterations were identified by serum levels. BM evaluation revealed small vacuoles with a distinct rounded shape and preferential cytoplasmic localization in promyelocytes, myelocytes (some of them with toxic granulation), erythroid precursors, and blasts (Figure 1B-C). Finally, both these patients had lower-risk MDS, characterized by normal cytogenetics and presence of the most common DNMT3A mutation (p.R882H), at a variant allele frequency of 26% in 1 of the 2 cases (supplemental Tables 1 and 2).
Of the remaining 7 cases, 4 had vacuolization of erythroid and/or myeloid precursors at onset, indicating that this morphologic feature may also be found in MNs with coexisting conditions. Indeed, although the diagnosis of a myeloid malignancy, especially MDS, may be sufficient for justifying the presence of vacuolization of HPs, all of these cases also had a diagnosis of an immune rheumatologic disorder (rheumatoid arthritis or ulcerative colitis), for which some patients were receiving active treatment (methotrexate), suggesting that the presence of coexisting conditions may determine these specific dysplastic features.
Lastly, the remaining 3 patients did not fulfill the diagnostic criteria for an MN, suggesting the vacuolization of HPs resulted from other biologic routes. In fact, the first patient was a 73-year-old woman with a history of alcohol abuse, secondary liver dysfunction, and copper deficiency. The second patient was an HIV+ 42-year-old man with celiac disease and secondary copper deficiency (Figure 1D), and the third was a 9-year-old boy with a complex congenital syndrome including neurologic, immunologic, and gastrointestinal disorders without evidence of copper and/or zinc alterations. More importantly, none of these 3 cases showed any cytogenetic abnormalities or any of the hallmark signs/symptoms associated with VEXAS syndrome.
The very low frequency of cells showing vacuoles in the BM smears of our cohort should be noted. For instance, vacuolization of HPs was reported at a median of 3.1% (range, 0.6% to 6.8% over 500 nucleated cells), regardless of the presence of a UBA1 mutation, further validating the rare and incidental nature of such findings (supplemental Figure 3). Of note, vacuoles were found to be negative on periodic acid–Schiff staining in both UBA1+ and UBA1− cases (supplemental Figure 4).
Cytoplasmic vacuoles in HPs can be seen in a number of clinical settings, including copper deficiency/zinc toxicity, alcohol abuse, antibiotic treatment, MDS, and VEXAS syndrome (supplemental Table 3).1,3,4 In the study by Beck et al,1 UBA1 mutations were identified in a cohort of 25 male patients with a late adult–onset (median age, 64 years) autoinflammatory syndrome characterized by fever, cytopenia, BM dysplastic features (eg, vacuolization of HPs, often in the context of MDS), neutrophilic dermatosis, joint pain, chondritis, and pulmonary inflammation. The presence of a hotspot mutation in UBA1 (p.Met41) resulted in a loss of function of the cytoplasmic isoform of UBA1, leading to immoderate production of the cytokines (interferon-γ, interleukin-8, and C-reactive protein) responsible for the associated autoinflammatory symptoms.1 In our case series, 2 patients tested positive for this mutation and showed the typical signs and symptoms of VEXAS syndrome, highlighting that testing for this mutation has to be taken into consideration in cases presenting with such clinical phenotypes. These 2 index cases also confirmed the association between UBA1, MDS, and Sweet syndrome and led us to review available BM biopsies of additional 4 Sweet syndrome cases and interrogate our collection of 200 exomes of MN patients. However, no other UBA1 mutations were found, and no vacuoles were observed. Notwithstanding, we do not exclude a strong association between Sweet syndrome, UBA1 mutation, and MDS, because this was also observed in the VEXAS report (8 of 25 patients with a mutation had Sweet syndrome, 2 of whom also had MDS).1
Furthermore, our case series also identified other conditions associated with vacuolization of HPs, such as copper deficiency, which encompasses neurologic and several hematologic findings (anemia, neutropenia, and rarely thrombocytopenia).2,7,8 Characteristic BM dysplasia in patients with copper deficiency includes, among others, vacuolization of erythroid and myeloid precursors, which may masquerade as MDS.2,8-11 In a recent study exploring the role of hypocupremia in patients with unexplained dysplastic cytopenia, only 1 patient registered low levels of this micronutrient, highlighting that indiscriminate testing may not be cost effective.2,12 However, the burden of an MDS misdiagnosis may be significant for both patients and clinicians, and therefore, it is mandatory to identify patients at risk for this rare condition, taking into consideration the presence of other disorders causing such peculiar morphologic features.
We acknowledge the limitations of the retrospective nature of this study with an informatics-based chart interrogation, which may not have accounted for cases with rare and therefore unreported vacuolated HPs. However, we showed that this feature is rarely found in BM specimens of patients undergoing evaluation for MNs and unexplained cytopenia. Nevertheless, if identified, this finding may help in clinical diagnosis by ruling out possible associated conditions.
Our results also emphasize that the morphologic evidence of vacuoles in blasts is associated with higher-risk MNs such as in cases of previous exposure to chemotherapy and/or radiation. Indeed, patients with a blast-only pattern of vacuolization had increased frequency of cytogenetic abnormalities. These alterations were linked to unique morphologic characteristics, such as cytoplasmic blast vacuolization, granulation, and blebbing in the case of high-risk MNs with chromothripsis.6 Moreover, blast vacuoles predicted poor overall survival in AML patients undergoing induction chemotherapy.5 Finally, it is noteworthy that various BM dysplastic changes have been previously found in patients receiving chemotherapy for hematologic malignancies; in the case of vacuolization of HPs, a putative treatment-related effect may be invoked, especially in cases where this feature appears in specimens collected during follow-up.13,14
In conclusion, our study demonstrates that vacuolization of HPs is an epiphenomenon shared by a variety of underlying disorders. This morphologic finding is not pathognomonic for any diagnosis, suggesting the need for a comprehensive clinical evaluation to guide physicians’ decisions and initial workup. Furthermore, the unique finding of vacuolization in a newly diagnosed patient with an MN should prompt the clinician to evaluate for cooccurring rheumatologic disorders and consider testing for UBA1 gene mutation if other causes have been excluded (zinc, copper, and alcoholism).
Requests for data sharing should be e-mailed to the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by the Edward P. Evans Foundation; the Henry & Marilyn Taub Foundation; the Aplastic Anemia and MDS International Foundation (grants R01HL118281, R01HL123904, R01HL132071, and R35HL135795) (J.P.M.); and a VeloSano Pilot Award and the Vera and Joseph Dresner Foundation–MDS (V.V.). C.G. was supported by the American-Italian Cancer Foundation Post-Doctoral Research Fellowship.
Authorship
Contribution: C.G., V.V., and J.P.M. generated and conceived the study design, figures, tables, and manuscript; C.G. and V.V. performed and analyzed sequencing data; H.J.R., L.D., and E.D.H. collected and analyzed hematopathologic data and provided detailed morphologic information; H.J.R. evaluated BM morphology and provided clinical expert input on the manuscript; S.P., H.A., S.K., L.T., M.Z., and H.E.C. reviewed the clinical data, took part in sample selection, and helped edit the manuscript; and all authors participated in data interpretation and critical review of the final paper and submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Valeria Visconte, Department of Translational Hematology and Oncology Research, Taussig Cancer Institute, 9620 Carnegie Ave, Building NE6-250, Cleveland, OH 44106; e-mail: visconv@ccf.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal