

In this issue of Blood, , by zooming in on the role of altered energy metabolism in the pathogenesis and chemoresistance of acute myeloid leukemia (AML) cells and leukemic stem cells (LSCs), implicate the enzyme very-long-chain acyl-CoA dehydrogenase (VLCAD) in supporting fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) in the mitochondrial metabolism of AML. They further demonstrate preclinical activity of a novel compound inhibiting VLCAD.1
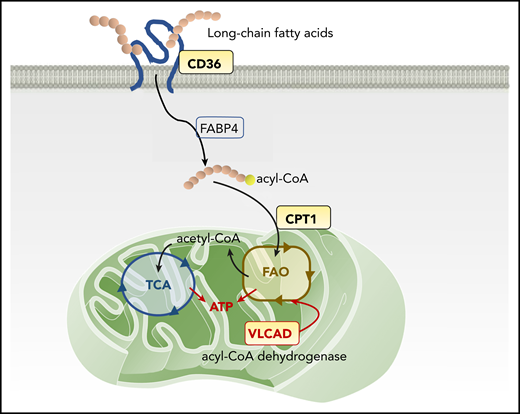
Mitochondrial FAO pathway and therapeutic targets. The scavenger receptor CD36 facilitates the uptake of long-chain FAs into the cytoplasm of AML cells. Inside the cells, FAs are first transported by FA-binding proteins (FABPs) and other transport proteins, followed by activation in a 2-step reaction: forming acyl-CoA in the cytoplasm and then FAO, forming acetyl-CoA inside mitochondria. CPT-1 conjugates FAs to carnitine, a prerequisite for mitochondrial translocation of FAs from the cytoplasm. In mitochondria, VLCAD, an intramitochondrial FAO enzyme, starts to catalyze the dehydrogenation reaction of long-chain FAs as the first intramitochondrial step of FAO. FAO generates ATP and provides acetyl-CoA to the tricarboxylic acid (TCA) cycle.
Mitochondrial FAO pathway and therapeutic targets. The scavenger receptor CD36 facilitates the uptake of long-chain FAs into the cytoplasm of AML cells. Inside the cells, FAs are first transported by FA-binding proteins (FABPs) and other transport proteins, followed by activation in a 2-step reaction: forming acyl-CoA in the cytoplasm and then FAO, forming acetyl-CoA inside mitochondria. CPT-1 conjugates FAs to carnitine, a prerequisite for mitochondrial translocation of FAs from the cytoplasm. In mitochondria, VLCAD, an intramitochondrial FAO enzyme, starts to catalyze the dehydrogenation reaction of long-chain FAs as the first intramitochondrial step of FAO. FAO generates ATP and provides acetyl-CoA to the tricarboxylic acid (TCA) cycle.
AML cells and LSCs are highly dependent on the production of mitochondrial biomass and rely on FAO2 and OXPHOS3 for survival. Compared with normal hematopoietic cells, they have a lower reserve capacity in their respiratory chain, which renders them susceptible to mitochondrial metabolic stress,4 a promising target for AML therapy. Emerging data highlight FAO as a key metabolic pathway fostering the survival of chemoresistant LSCs. LSCs from patients with relapsed AML acquire a compensatory ability to overcome the loss of amino acid metabolism by increasing FAO.5 This mechanism was recently implicated as causing resistance to azacitidine/venetoclax therapy, a widely used induction regimen for elderly patients with AML.6 In preclinical models, cytarabine arabinoside (AraC)–resistant AML cells displayed increased FAO and OXPHOS, and the FAO inhibitor etomoxir induced an energy shift from high to low OXPHOS, which sensitized these cells to AraC.3 However, the molecular mechanisms of FAO activation in AML cells remained unknown. Thus, the identification of VLCAD by Tcheng et al offers a novel, potentially druggable therapeutic target that is highly selective for AML cells and could be safely translated into clinical trials.
Mitochondrial FAO, the primary catabolic pathway for lipids, generates the reducing compounds NADH and FADH-2 for the electron transport chain and provides acetyl-CoA to the tricarboxylic acid cycle to produce adenosine triphosphate (ATP; see figure). The first step in cellular FA utilization is the uptake of long-chain FAs into the cytoplasm, a process facilitated by the scavenger receptor CD36, fatty acid–binding proteins, and transport proteins.7 FAs are then activated in a 2-step reaction, first forming acyl-CoA in the cytoplasm and then breaking down into acetyl-CoA via FAO inside the mitochondria. A rate-limiting step of FAO is catalyzed by carnitine palmitoyl transferase-1 (CPT-1), which conjugates FAs to carnitine, a prerequisite for mitochondrial translocation of FAs from the cytoplasm.7 These FA uptake and consumption mechanisms affect the fate of LSCs, in particular their adaptation to a specialized bone marrow microenvironment and response/resistance to drugs.8 Thus, CD36 and CPT-1 have been considered potential pharmacological targets for FAO inhibition in AML. A CD36 neutralizing antibody has been shown to impair metastasis of human melanoma and breast cancer cells.9 Inhibition of CPT-1 causes mitochondrial damage and induces cell death in primary AML cells.10
The intramitochondrial FAO enzyme VLCAD, identified by Tcheng et al, catalyzes the first intramitochondrial step of long-chain FAOs. They further identified a polyhydroxylated fatty alcohol with a terminal alkyne known as avocadyne (16-heptadecyne-1,2,4-triol; AYNE) as a potent small-molecule VLCAD inhibitor in a respirometry-based screening. By using both VLCAD knockdown and the novel pharmacological inhibitor AYNE, the research team convincingly demonstrated reduction of mitochondrial respiration caused by FAO alteration, leading to reduced ATP production in AML cells, despite moderate upregulation of glycolysis, and to decreased AML cell viability and proliferation. These results are supported by multiple orthogonal assays, such as intricate respiration assays supplying FAs as a source of fuel, enzymatic assays, and metabolomic analyses. Notably, normal hematopoietic stem cells (HSCs) compensate for this through glycolytic processes, thereby maintaining their ATP levels and viability. Thus, this finding is consistent with the notion that AML cells, but not HSCs, are metabolically dependent on FAO and OXPHOS for survival. In a mouse engraftment assay, pharmacological inhibition of VLCAD with AYNE was further tested in vivo in a functionally defined subset of primitive human AML and normal hematopoietic cell populations. The results showed that 6 weeks of AYNE therapy was well tolerated and that VLCAD inhibition significantly reduced the repopulation potential of leukemia cells.
The discovery of VLCAD as a novel, potentially druggable target in AML cells emphasizes the role of mitochondrial metabolism in AML and prompts further in-depth exploration of the metabolic dependencies of AML and their potential for therapeutic translation. Several important questions await further study. Are all AMLs FAO dependent, or is this phenomenon genotype conferred? Is a therapeutic window achievable with chronic FAO inhibition, given cardiomyopathy concerns that arose in acute murine deletion experiments, or is intermittent blockade of the pathway sufficient to induce death of malignant cells and spare normal cells? Given the heterogeneity and multiclonal nature of AML, would blocking just one arm of the multifaceted metabolic machinery produce antileukemia efficacy before metabolic adaptation? Or should the focus shift toward targeting the residual AML cells and LSCs that survive chemotherapeutic stress and have been shown to have specific metabolic vulnerabilities?
Despite these unknowns, Tcheng et al have aided the field by providing a previously missing link in our understanding of how AML subverts the cell’s powerful energy-generating machinery to its favor, thereby creating a metabolic Achilles heel that can be exploited therapeutically. Their comprehensive biochemical and mechanistic analyses targeting VLCAD in FAO offer the possibility of future combinatorial therapeutic strategies that could facilitate the elimination of AML cells and LSCs while reducing on-target, off-tumor toxicity.
Conflict-of-interest disclosure: Y.T. reports grants and other support from Sysmex, Kohjin Bio, and Roche Diagnostics. M.K. reports grants and other support outside the submitted work from AbbVie, F. Hoffman-La Roche, Stemline Therapeutics, Forty-Seven, and Genentech; grants outside the submitted work from Eli Lilly, Cellectis, Calithera, Ablynx, Agios, Ascentage, AstraZeneca, Rafael Pharmaceutical, and Sanofi; and other support from Reata Pharmaceuticals and Janssen. In addition, M.K. has patents US 7,795,305 B2 “CDDO-compounds and combination therapies thereof” with royalties paid to Reata Pharmaceuticals; “Combination therapy with a mutant IDH1 inhibitor and a BCL-2” licensed to Eli Lilly, and 62/993,166 “Combination of a MCL-1 inhibitor and midostaurin, uses and pharmaceutical compositions thereof” pending to Novartis.
