

Key Points
TP53 loss impairs BAX/BAK activation, conferring a competitive advantage to prolonged and sublethal targeting of either BCL-2 or MCL-1.
Simultaneous targeting of BCL-2 and MCL-1 increases the early apoptotic response in TP-53–deficient cells, enhancing long-term outcomes.

Abstract
Selective targeting of BCL-2 with the BH3-mimetic venetoclax has been a transformative treatment for patients with various leukemias. TP-53 controls apoptosis upstream of where BCL-2 and its prosurvival relatives, such as MCL-1, act. Therefore, targeting these prosurvival proteins could trigger apoptosis across diverse blood cancers, irrespective of TP53 mutation status. Indeed, targeting BCL-2 has produced clinically relevant responses in blood cancers with aberrant TP-53. However, in our study, TP53-mutated or -deficient myeloid and lymphoid leukemias outcompeted isogenic controls with intact TP-53, unless sufficient concentrations of BH3-mimetics targeting BCL-2 or MCL-1 were applied. Strikingly, tumor cells with TP-53 dysfunction escaped and thrived over time if inhibition of BCL-2 or MCL-1 was sublethal, in part because of an increased threshold for BAX/BAK activation in these cells. Our study revealed the key role of TP-53 in shaping long-term responses to BH3-mimetic drugs and reconciled the disparate pattern of initial clinical response to venetoclax, followed by subsequent treatment failure among patients with TP53-mutant chronic lymphocytic leukemia or acute myeloid leukemia. In contrast to BH3-mimetics targeting just BCL-2 or MCL-1 at doses that are individually sublethal, a combined BH3-mimetic approach targeting both prosurvival proteins enhanced lethality and durably suppressed the leukemia burden, regardless of TP53 mutation status. Our findings highlight the importance of using sufficiently lethal treatment strategies to maximize outcomes of patients with TP53-mutant disease. In addition, our findings caution against use of sublethal BH3-mimetic drug regimens that may enhance the risk of disease progression driven by emergent TP53-mutant clones.
Introduction
Venetoclax has emerged as a new and effective treatment option for patients with chronic lymphocytic leukemia (CLL)1 or acute myeloid leukemia (AML).2-4 Venetoclax belongs to a novel BH3-mimetic class of small molecules that selectively target BCL-2, MCL-1, or their prosurvival relatives, activating the apoptosis effectors BAX and BAK to drive mitochondrial outer membrane permeabilization (MOMP) and cell death.5
The tumor suppressor TP-53 coordinates the cellular response to noxious stress, including DNA damage. TP-53 activation leads to increased expression of genes encoding BH3-only proteins (PUMA, NOXA) that promote apoptosis. TP-53 operates upstream of the BCL-2 protein family, supporting the notion that defects in TP-53 activity should not compromise the action of BH3-mimetic drugs.5 Consistent with this, TP53-mutant CLL cells are highly sensitive to venetoclax in vitro,6 and clinical responses to venetoclax are similar for both wild-type and TP53-mutant CLL,1,6 in contrast to inferior outcomes of conventional chemotherapy in patients with TP53-mutant disease. However, recent studies challenge the assumption that BH3-mimetic activity is agnostic to TP-53 status. Genetic screening in AML cell lines has linked venetoclax resistance to TP-53 loss.7-9 In clinical studies, despite initial responses to venetoclax, TP-53 abnormalities predispose patients to earlier disease recurrence of CLL10 or AML, despite combination with chemotherapy or hypomethylating agents.3,11,12
To reconcile these apparently contradictory observations, we investigated the impact of TP-53 dysfunction on response of hematological malignancies to BH3-mimetic drugs. Across multiple models and corroborated by clinical observations, we observed that, in the presence of a sublethal concentration of BH3-mimetic drugs targeting either BCL-2 or MCL-1, TP-53 deficiency conferred a competitive advantage to surviving cells, impairing activation of proapoptotic BAX/BAK and enabling cells to outgrow their TP-53–intact counterparts. Although combining BH3-mimetics with cytotoxic drugs failed to avert outgrowth of TP-53–deficient cells, sustained responses could be achieved when BH3-mimetic agents were combined to cotarget BCL-2 and MCL-1, resulting in potent activity, irrespective of TP53 mutation status.
Our findings have significant implications for how BH3-mimetic drugs are ideally used in the clinic. Our results highlight the role of defective TP-53 in promoting treatment failure if a sublethal BH3-mimetic dose is used. Furthermore, our results support the deployment of novel therapeutic approaches with TP-53–independent activity, such as a tandem BH3-mimetic strategy to improve treatment outcomes in leukemias with defective TP-53.
Methods
Full details are provided in supplemental Materials, available on the Blood Web site.
Primary human AML specimens
Samples were obtained after the patients provided informed consent. Bone marrow (BM) samples were from patients participating in the phase 1 dose-escalation study of the BCL-2 inhibitor S55746 as a monotherapy in AML and higher risk myelodysplasia (Protocol CL1-055746-002). Additional samples were collected from patients participating in the Chemotherapy and Venetoclax in Elderly AML Trial (CAVEAT).12 All experiments were conducted in accordance with institutional requirements and the Declaration of Helsinki.
Mice
Mouse experiments were conducted with institutional approvals. Cells (4 × 105 MOLM-13; 1 × 105 MV-4-11) were injected IV into nonirradiated NOD-SCID IL2Rγ-null (NSG) mice. Primary AML leukemic blasts (1 × 106) were injected into NOD-IL2Rcγnull (NRG-SG3) mice. Mice were treated with various dose regimens of the BH3-mimetics targeting BCL-2 and MCL-1.
CRISPR/Cas9 technology
Virally delivered CRISPR/Cas9 technology was used to derive gene KO variants of diverse blood cancer–derived cell lines.13 Whole-genome KO screenings to identify resistance factors to the MCL-1 inhibitor (MCL-1i) S63845 were performed by transducing Cas9-expressing Eµ-Myc lymphoma cells with the mouse YUSA library.14
In vitro cell growth competition assays on human blood cancer–derived cell lines
Control WT TP53 GFP+ cells were seeded at various ratios with test KO TP53 BFP+ cells and treated with doses of the BH3-mimetics or dimethyl sulfoxide (DMSO) as the control. The relative proportion of viable GFP+ or BFP+ cells was evaluated by flow cytometry (CytoFLEX or LSR II; BD). Similar experiments were performed with Eµ-Myc mouse lymphoma cells.
Measuring BAX and BAK activation by flow cytometry
BH3-mimetic (or DMSO)–treated human cancer cells were fixed, permeabilized, and stained with antibodies to activated BAX (biotinylated rat monoclonal clone 1E5; Walter and Eliza Hall Institute), activated BAK (mouse monoclonal clone G317-2; cat. no. 556382; BD Biosciences) and antibodies against cytochrome c (clone REA702, cat. no. 130-111-368; Miltenyi Biotec). Cells were analyzed on a FACSFortessa (BD Biosciences).
Results
Striking clinical response of a TP53-null AML to BCL-2 inhibition
We initially examined our records for patients with TP53-aberrant AML who had been treated with only BCL-2 inhibition. A patient with monosomal karyotype (45,XX,add(11)(p12),-17) AML refractory to standard induction chemotherapy received S55746,15,16 a selective BCL-2 inhibitor equivalent to venetoclax.17 At screening, the leukemia harbored a dominant TP53-null clone (monosomy 17 and a P151H mutation). After S55746 for 21 days, the patient achieved near-complete remission with marked reduction in the variant allele burden of the TP53-mutated clone, along with concordant reduction in IDH1 mutation (Figure 1A; supplemental Table 1). Two further courses of S55746 were administered before the patient underwent an elective allogeneic hematopoietic stem cell transplant.
![Selection for clones harboring mutant TP53 during submaximal venetoclax treatment in some human patients with primary AML. (A) Rapid leukemic clearance in a patient with biallelic TP53-aberrant AML (monosomy 17 and pathogenic TP53 mutation; also carrying an IDH1 R132C mutation, but negative for FLT3-ITD, FLT3-TKD, NPM1, or CEBPa mutations) after treatment with the BCL-2 inhibitor S55746. Although the leukemia in this patient was refractory to 2 cycles of standard AML induction chemotherapy (cytarabine+idarubicin; with the second cycle incorporating high-dose cytarabine) with blasts comprising 70% BM cells, it rapidly responded after administration of a single 21-day cycle of 100 mg/d of the oral BCL-2 inhibitor S55746, achieving morphological remission (4th column from left). Sustained reduction in the leukemic clone was confirmed by cytogenetics and whole-exome sequencing during ongoing BCL-2i therapy bridging to allogeneic BM transplantation (7th column from left) (variant allele frequency [VAF]: TP53 refers to NM_001126112.2:c.452C>A, and IDH1 refers to NM_005896.2:c.394C>T). (B) BM blast changes among patients exposed to 7 days of venetoclax monotherapy stratified by TP53 status. The median blast reductions for TP53 WT (n = 31) or TP53 mutant cases (n = 9) are represented by a red line. Open circles, TP53 mutation VAFs >50%; open squares, VAFs <50%. (C-F) Single-cell DNA sequencing analyses using a Mission Bio Tapestri instrument of BM (CAL-012, CAL-030, and CVC-001) or peripheral blood (PB; CAU-001) samples from 4 patients with AML. The bar graphs indicate the proportion of TP53 wild-type (in gray) and TP53-mutant (red/blue) clones in each sample during venetoclax treatment (CAL-012 [C]; CAL-030 [D]; CVC-001 [E]; and CAU-001 [F]). TP53 status, as well as the commutations (N-RAS, K-RAS, and PTPN11) present in TP53-mutated subclones (blue) are also indicated.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020010167/2/m_bloodbld2020010167f1.png?Expires=1763476112&Signature=DSKkRKzQY791yti7VbS6Zwu3PdVhG93V6Dtc2WKSGVkzGplpPt9uyW3pGIclx8faLEJxKKdn4hkM44J2ASmO-XoUJnihI8IKTVhmJDYcQSuWarkrdQ-48I61kOMTWHKyR4ZyvY8fMfc8mHi9MioGpcYteFlOq~b6q0pqo0-o1I8DhPu3BZ6GglZhTgVVEkjvUaigt-H5zFCB5t4zzYG80H1xL6AhQSGaYej-3g5Wq43GvfCI5Za~5xWW-J2gx4JTbarfCJgmgJCXsp4OFXElQO~2DiOwmCiyOwxu1-tIugkQR6AMrBxNRYpRakrXgl7rVcRr7qbZ-xElWe8I-hY~aA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Selection for clones harboring mutant TP53 during submaximal venetoclax treatment in some human patients with primary AML. (A) Rapid leukemic clearance in a patient with biallelic TP53-aberrant AML (monosomy 17 and pathogenic TP53 mutation; also carrying an IDH1 R132C mutation, but negative for FLT3-ITD, FLT3-TKD, NPM1, or CEBPa mutations) after treatment with the BCL-2 inhibitor S55746. Although the leukemia in this patient was refractory to 2 cycles of standard AML induction chemotherapy (cytarabine+idarubicin; with the second cycle incorporating high-dose cytarabine) with blasts comprising 70% BM cells, it rapidly responded after administration of a single 21-day cycle of 100 mg/d of the oral BCL-2 inhibitor S55746, achieving morphological remission (4th column from left). Sustained reduction in the leukemic clone was confirmed by cytogenetics and whole-exome sequencing during ongoing BCL-2i therapy bridging to allogeneic BM transplantation (7th column from left) (variant allele frequency [VAF]: TP53 refers to NM_001126112.2:c.452C>A, and IDH1 refers to NM_005896.2:c.394C>T). (B) BM blast changes among patients exposed to 7 days of venetoclax monotherapy stratified by TP53 status. The median blast reductions for TP53 WT (n = 31) or TP53 mutant cases (n = 9) are represented by a red line. Open circles, TP53 mutation VAFs >50%; open squares, VAFs <50%. (C-F) Single-cell DNA sequencing analyses using a Mission Bio Tapestri instrument of BM (CAL-012, CAL-030, and CVC-001) or peripheral blood (PB; CAU-001) samples from 4 patients with AML. The bar graphs indicate the proportion of TP53 wild-type (in gray) and TP53-mutant (red/blue) clones in each sample during venetoclax treatment (CAL-012 [C]; CAL-030 [D]; CVC-001 [E]; and CAU-001 [F]). TP53 status, as well as the commutations (N-RAS, K-RAS, and PTPN11) present in TP53-mutated subclones (blue) are also indicated.
Selection for clones harboring mutant TP53 during submaximal venetoclax treatment in some human patients with primary AML. (A) Rapid leukemic clearance in a patient with biallelic TP53-aberrant AML (monosomy 17 and pathogenic TP53 mutation; also carrying an IDH1 R132C mutation, but negative for FLT3-ITD, FLT3-TKD, NPM1, or CEBPa mutations) after treatment with the BCL-2 inhibitor S55746. Although the leukemia in this patient was refractory to 2 cycles of standard AML induction chemotherapy (cytarabine+idarubicin; with the second cycle incorporating high-dose cytarabine) with blasts comprising 70% BM cells, it rapidly responded after administration of a single 21-day cycle of 100 mg/d of the oral BCL-2 inhibitor S55746, achieving morphological remission (4th column from left). Sustained reduction in the leukemic clone was confirmed by cytogenetics and whole-exome sequencing during ongoing BCL-2i therapy bridging to allogeneic BM transplantation (7th column from left) (variant allele frequency [VAF]: TP53 refers to NM_001126112.2:c.452C>A, and IDH1 refers to NM_005896.2:c.394C>T). (B) BM blast changes among patients exposed to 7 days of venetoclax monotherapy stratified by TP53 status. The median blast reductions for TP53 WT (n = 31) or TP53 mutant cases (n = 9) are represented by a red line. Open circles, TP53 mutation VAFs >50%; open squares, VAFs <50%. (C-F) Single-cell DNA sequencing analyses using a Mission Bio Tapestri instrument of BM (CAL-012, CAL-030, and CVC-001) or peripheral blood (PB; CAU-001) samples from 4 patients with AML. The bar graphs indicate the proportion of TP53 wild-type (in gray) and TP53-mutant (red/blue) clones in each sample during venetoclax treatment (CAL-012 [C]; CAL-030 [D]; CVC-001 [E]; and CAU-001 [F]). TP53 status, as well as the commutations (N-RAS, K-RAS, and PTPN11) present in TP53-mutated subclones (blue) are also indicated.
Analogous to the response to venetoclax in TP53-mutant CLL,1,6 this AML case illustrates that defective TP53 does not preclude a robust clinical response to BCL-2 inhibition. This prompted us to further explore BCL-2 inhibition in patients with TP53 abnormalities.
TP53-mutant AML clones expand on short-term venetoclax exposure
To gain further insight into the clinical impact of venetoclax on TP53-mutated AML, we compared the leukemic burden in BM before and after a 7-day prephase of venetoclax, with doses ranging from 50 to 600 mg/d (maximum dose approved in AML), given before the addition of cytarabine and idarubicin as part of a phase 1b clinical trial (CAVEAT).12 Nine patients (supplemental Table 2) harboring TP53-mutant AML, displayed a variable response to venetoclax (Figure 1B). Although BM blast reductions were noted in some patients, we focused attention on 4 venetoclax nonresponsive cases. Paired BM samples before and after 7 days of venetoclax were characterized by single-cell targeted DNA sequencing (supplemental Table 3). In all 4 cases, expansion of TP53-mutant relative to TP53-wild-type clones was noted, even at the highest venetoclax dose (600 mg; Figure 1C-F). In 2 patients (CAL-012 and CAL-030; Figure 1C-D), expansion of TP53-mutant clones was propagated by commutations known to promote cell growth (N-RAS, K-RAS, and PTPN11). Therefore, although prior reports suggest that TP-53 deletion in AML cell lines is associated with venetoclax resistance in vitro,8 our clinical findings suggest that early response of TP53 mutant AML clones to venetoclax may be variable.
TP53 loss variably impairs the induction of apoptosis by venetoclax
To explore the consequences of TP53 deficiency on response to BH3-mimetic drugs, TP53 was deleted by CRISPR/Cas9 in 2 human AML cell lines: MOLM-13 and MV-4-11 (Figure 2A; supplemental Figure 1A). As expected, TP53 loss rendered both cell lines resistant to apoptosis by an MDM2 inhibitor (supplemental Figure 1B). In contrast, the impact of TP-53 loss on venetoclax sensitivity in short-term (24 hour) assays was variable, with the 50% inhibitory concentration (IC50) increased by approximately twelvefold to eightfold in MV-4-11 cells (supplemental Figure 1C) to only approximately threefold in MOLM-13 cells (Figure 2B). These results prompted further exploration into how TP-53 loss may influence the response to venetoclax.
![TP53 loss enables AML cells to rapidly escape suboptimal BCL-2 inhibition. (A) Western blot analysis confirming CRISPR/Cas9-induced loss of TP-53 expression in 2 independent clones of the MOLM-13 human AML derived cell line, expressing guide RNAs to target TP53. In all cases, cells were cotreated with an MDM2 inhibitor for 16 hours to induce expression and activation of TP-53, as well as the broad-spectrum caspase inhibitor Q-VD-OpH to prevent late stages of apoptosis that are associated with protein degradation. (B) Wild-type (WT) or TP53-deficient (KO) clones of MOLM-13 were treated with 0 to 10 μM venetoclax, and their viability was determined 24 hours later. (C) Schematic of the in vitro cell competition assay used to evaluate the impact of suboptimal venetoclax treatment over several weeks on TP53 WT and isogenic KO clones. Equivalent (50:50) numbers of TP53 WT (GFP+) and TP53 KO (BFP+) cells were seeded and cocultured in the continued presence of the indicated doses of venetoclax. Cell survival was monitored by flow cytometry to track GFP+ and BFP+ cells in viable PI− cells. (D) The growth of WT or TP53 KO MOLM-13 cells treated continuously with a suboptimal (IC20) dose (supplemental Table 4) of venetoclax (or DMSO) was monitored, as in panel C. (E) The outgrowth of TP53 KO MOLM-13 cells seeded in a 5:95 TP53 KO/TP53 WT ratio treated continuously with an IC50 dose of venetoclax (or control DMSO treated) was monitored by flow cytometric analysis. Data are means ± SD of ≥3 independent experiments. (F) The viability of aliquots of TP53 KO (right) MOLM-13 cells or their TP53 WT controls (EV−, empty vector; left) maintained continuously in IC20 dose of venetoclax for 0, 7, 14, 21, and 28 days (see panel D) treated for 24 hours with 0 to 10 μM venetoclax was determined as in panel B; the IC50 values are detailed in supplemental Table 5. (G) WT MOLM-13 and TP53 KO cells were transplanted at a 50:50 ratio into NOD SCID IL2Rγ−/− (NSG) mice. Three days after transplantation, the mice were treated with vehicle, or venetoclax (75 mg/kg by oral gavage once daily [QD] on week days) for 2 weeks. (H) NSG mice were transplanted with equal numbers of TP53 WT and TP53 KO MOLM-13 cells. Three days afterward, mice were treated with vehicle or venetoclax as shown in panel G. The proportions of TP53 WT (GFP+) and KO (BFP+) human (CD45+) cells in the peripheral blood were determined by flow cytometry after the 2-week treatment course. ***P = 5.5 × 10−5 vehicle vs venetoclax treatment of TP53 KO cells. Data are means ± SD of 5 animals per group from a representative experiment (n = 3 independent experiments).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020010167/2/m_bloodbld2020010167f2.png?Expires=1763476112&Signature=2jrxzzr9FGk5eg~L9o1Hojuzbj4yrhk~7apXNAJxcyCLwPI1p7pCQzBLRq4wTO3T4CfOaPfNSL7SFopIRHKneKxcAupCh46HXU5zlgSfioxT6EM3dja6MV0F2dAQ6G7PwB-u2M97Moy-mKC~d5V27ORIBIZQ5Y-LcE29g2QrmJOCurXJf0MLZqOXgsuEFei59n8nkPJU~EMUAHFgzDKc7M-Ua-ivMAOAQrFuVQiam6m8qF5P0XDux9D20hB6oUwTkgHJq2hVN5Ql1xNatz97d17JczXvErB57mcX8ws8LJNGCYfdLr6~ASNrnpz5DpVY3AtSqqdPDD3NLjm9Fc7dEQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TP53 loss enables AML cells to rapidly escape suboptimal BCL-2 inhibition. (A) Western blot analysis confirming CRISPR/Cas9-induced loss of TP-53 expression in 2 independent clones of the MOLM-13 human AML derived cell line, expressing guide RNAs to target TP53. In all cases, cells were cotreated with an MDM2 inhibitor for 16 hours to induce expression and activation of TP-53, as well as the broad-spectrum caspase inhibitor Q-VD-OpH to prevent late stages of apoptosis that are associated with protein degradation. (B) Wild-type (WT) or TP53-deficient (KO) clones of MOLM-13 were treated with 0 to 10 μM venetoclax, and their viability was determined 24 hours later. (C) Schematic of the in vitro cell competition assay used to evaluate the impact of suboptimal venetoclax treatment over several weeks on TP53 WT and isogenic KO clones. Equivalent (50:50) numbers of TP53 WT (GFP+) and TP53 KO (BFP+) cells were seeded and cocultured in the continued presence of the indicated doses of venetoclax. Cell survival was monitored by flow cytometry to track GFP+ and BFP+ cells in viable PI− cells. (D) The growth of WT or TP53 KO MOLM-13 cells treated continuously with a suboptimal (IC20) dose (supplemental Table 4) of venetoclax (or DMSO) was monitored, as in panel C. (E) The outgrowth of TP53 KO MOLM-13 cells seeded in a 5:95 TP53 KO/TP53 WT ratio treated continuously with an IC50 dose of venetoclax (or control DMSO treated) was monitored by flow cytometric analysis. Data are means ± SD of ≥3 independent experiments. (F) The viability of aliquots of TP53 KO (right) MOLM-13 cells or their TP53 WT controls (EV−, empty vector; left) maintained continuously in IC20 dose of venetoclax for 0, 7, 14, 21, and 28 days (see panel D) treated for 24 hours with 0 to 10 μM venetoclax was determined as in panel B; the IC50 values are detailed in supplemental Table 5. (G) WT MOLM-13 and TP53 KO cells were transplanted at a 50:50 ratio into NOD SCID IL2Rγ−/− (NSG) mice. Three days after transplantation, the mice were treated with vehicle, or venetoclax (75 mg/kg by oral gavage once daily [QD] on week days) for 2 weeks. (H) NSG mice were transplanted with equal numbers of TP53 WT and TP53 KO MOLM-13 cells. Three days afterward, mice were treated with vehicle or venetoclax as shown in panel G. The proportions of TP53 WT (GFP+) and KO (BFP+) human (CD45+) cells in the peripheral blood were determined by flow cytometry after the 2-week treatment course. ***P = 5.5 × 10−5 vehicle vs venetoclax treatment of TP53 KO cells. Data are means ± SD of 5 animals per group from a representative experiment (n = 3 independent experiments).
TP53 loss enables AML cells to rapidly escape suboptimal BCL-2 inhibition. (A) Western blot analysis confirming CRISPR/Cas9-induced loss of TP-53 expression in 2 independent clones of the MOLM-13 human AML derived cell line, expressing guide RNAs to target TP53. In all cases, cells were cotreated with an MDM2 inhibitor for 16 hours to induce expression and activation of TP-53, as well as the broad-spectrum caspase inhibitor Q-VD-OpH to prevent late stages of apoptosis that are associated with protein degradation. (B) Wild-type (WT) or TP53-deficient (KO) clones of MOLM-13 were treated with 0 to 10 μM venetoclax, and their viability was determined 24 hours later. (C) Schematic of the in vitro cell competition assay used to evaluate the impact of suboptimal venetoclax treatment over several weeks on TP53 WT and isogenic KO clones. Equivalent (50:50) numbers of TP53 WT (GFP+) and TP53 KO (BFP+) cells were seeded and cocultured in the continued presence of the indicated doses of venetoclax. Cell survival was monitored by flow cytometry to track GFP+ and BFP+ cells in viable PI− cells. (D) The growth of WT or TP53 KO MOLM-13 cells treated continuously with a suboptimal (IC20) dose (supplemental Table 4) of venetoclax (or DMSO) was monitored, as in panel C. (E) The outgrowth of TP53 KO MOLM-13 cells seeded in a 5:95 TP53 KO/TP53 WT ratio treated continuously with an IC50 dose of venetoclax (or control DMSO treated) was monitored by flow cytometric analysis. Data are means ± SD of ≥3 independent experiments. (F) The viability of aliquots of TP53 KO (right) MOLM-13 cells or their TP53 WT controls (EV−, empty vector; left) maintained continuously in IC20 dose of venetoclax for 0, 7, 14, 21, and 28 days (see panel D) treated for 24 hours with 0 to 10 μM venetoclax was determined as in panel B; the IC50 values are detailed in supplemental Table 5. (G) WT MOLM-13 and TP53 KO cells were transplanted at a 50:50 ratio into NOD SCID IL2Rγ−/− (NSG) mice. Three days after transplantation, the mice were treated with vehicle, or venetoclax (75 mg/kg by oral gavage once daily [QD] on week days) for 2 weeks. (H) NSG mice were transplanted with equal numbers of TP53 WT and TP53 KO MOLM-13 cells. Three days afterward, mice were treated with vehicle or venetoclax as shown in panel G. The proportions of TP53 WT (GFP+) and KO (BFP+) human (CD45+) cells in the peripheral blood were determined by flow cytometry after the 2-week treatment course. ***P = 5.5 × 10−5 vehicle vs venetoclax treatment of TP53 KO cells. Data are means ± SD of 5 animals per group from a representative experiment (n = 3 independent experiments).
Prolonged exposure to sublethal venetoclax treatment promotes outgrowth of TP53-deficient AML
As resistance to venetoclax imparted by TP-53 loss in short-term assays was only modest, we next examined the impact of TP53 loss in longer term assays spanning several weeks. Fluorescently marked TP53-wild-type (GFP+) and TP53-deficient (BFP+) MOLM-13 cells were mixed in equal ratio and their proportions tracked during continuous exposure to venetoclax (Figure 2C). Although TP53-deletion did not alter outcomes using an IC80 dose of venetoclax, with cells of either genotype killed within 48 hours (not shown), an IC20 of venetoclax led to marked outgrowth of TP53-deficient relative to TP53-wild-type cells after 1 week of therapy, whereas spontaneous outgrowth of TP53-deficient cells in control media was more gradual (Figure 2D). Outgrowth of TP53-deficient over wild-type cells seeded at a 1:20 ratio was also evident using an IC50 of venetoclax (Figure 2E). Collectively, these results suggest that short-term measures of venetoclax drug sensitivity (Figure 2B) may conceal the potential for dominant outgrowth of TP53-deficient cells exposed to suboptimal concentrations over an extended period (Figure 2D-E). Short-term cytotoxicity experiments performed weekly during the long-term cell competition assays revealed no significant change in the IC50 sensitivity of TP53-deficient MOLM-13 cells to venetoclax, thus ruling out the possibility of acquired drug resistance (Figure 2F).
To determine the relevance of these findings in vivo, an equal ratio of TP53-wild-type and -deficient MOLM-13 cells were inoculated into NSG mice, and 3 days later, treatment with either vehicle or venetoclax (75 mg/kg per day) for a 2-week period commenced (Figure 2G). By the completion of treatment, the proportion of TP53-deficient to wild-type MOLM-13 cells was significantly higher in the venetoclax-treated cohort (Figure 2H). These findings confirm the potential for venetoclax to favor expansion of TP53-deficient, compared with wild-type AML populations in vivo (Figure 1C-F).
Similar findings were made with TP53-deficient MV-4-11 cells.11 Moreover, MV-4-11 cells harboring a spontaneous TP53 mutation (R248W) also exhibited a competitive advantage over wild-type counterparts when exposed to a sublethal IC20 of venetoclax in extended cultures (supplemental Figure 1D). Collectively, these findings reveal a competitive advantage for TP-53–defective AML populations if treated for extended periods with a suboptimal concentration of venetoclax.
Suboptimal doses of venetoclax enhance the competitive fitness of TP53-deficient B-cell malignancies
We extended our experiments to determine whether defective TP53 could present a barrier to venetoclax efficacy in models of B-cell malignancy. Deleting TP53 in the human B-ALL cell line RS4;11 (Figure 3A) impaired killing by an MDM2 inhibitor (supplemental Figure 2A), but had no impact on venetoclax sensitivity in short-term (24 hour) assays (Figure 3B). However, in extended duration cell competition assays, TP53-deficient RS4;11 cells expanded more rapidly during continuous exposure to sublethal vs higher doses of venetoclax (Figure 3C-D). Furthermore, there was no evidence of acquired increased drug resistance during the 4-week culture period (Figure 3E). Similar findings were observed for the diffuse large B-cell lymphoma (DLBCL) cell line OCI-LY19 (Figure 3F; supplemental Figure 2B).
![TP53 loss enables blood cancer cells to rapidly escape suboptimal BCL-2 inhibition. (A) Western blot confirming CRISPR/Cas9-induced loss of TP-53 expression in independent clones of the RS4;11 human ALL-derived cell line, expressing sgRNAs to target TP53. In all cases, cells were cotreated with an MDM2 inhibitor for 16 hours to induce expression and activation of TP-53, as well as the broad-spectrum caspase inhibitor Q-VD-OpH to prevent late stages of apoptosis that are associated with protein degradation. (B) TP53 wild-type (WT) or TP53-deficient (KO) clones of RS4;11 were treated with 0 to 10 μM venetoclax and their viability determined 24 hours later; the IC50 values are indicated in parentheses. (C) The growth of WT or TP53 KO RS4;11 cells treated continuously with a suboptimal (IC20) dose (supplemental Table 4) of venetoclax (or control treated) was monitored. (D) The outgrowth of TP53 KO RS4;11 cells seeded in a 5 TP53 KO:95 TP53 WT ratio treated continuously with an IC50 dose of venetoclax (or control treated) was monitored by flow cytometric analysis. Data are means ± SD of ≥3 independent experiments. (E) The viability of aliquots of TP53 KO RS4;11 cells or their TP53 WT controls (EV− empty vector) maintained continuously in an IC20 dose of venetoclax for 0, 7, 14, 21, and 28 days (see panel C) treated for 24 hours with 0 to 10 μM venetoclax was determined as in panel B; the IC50 values are indicated in supplemental Table 5. (F) Experiments similar to those with RS4;11 ALL cells (A-B, D) were undertaken with OCI-LY19 cells, a human DLBCL-derived cell line. Data from cell viability (Cell-Titer Glo) or cell competition (fluorescence-activated cell sorting [FACS]) assays represent means ± SD of ≥3 independent experiments.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020010167/2/m_bloodbld2020010167f3.png?Expires=1763476112&Signature=T4sfDEx-AKST0B6MFtG07DshvFHYn1hx-k33iDZD~y30yjVbQY~XRtMLB~84UPfxrbiLjHgeUpFNJpAPqgqRhEGmEYKW1X2oJuWLofHKglui5GZ1lzNFDNd4DpGWhMeAk0qCx~QF3aN97upjsHspDAJffPyiH1nW5gDNb6Z~JnvHdIm~ozLE~HJxlzk7kJxeSzX-u-R2S1RxcvrXfi8kmksHn1FdiNjycSfc4J4D69gOn-ik3U7q2Kayq1noXIMNvxTVI6RknhlusH0ymUfSSdl-eNVVbgejWVTwLqGpTK~bm9c6yJsr2LpYYU0hrGKUqmP6bX25ZFpR2kv0Ze58kA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TP53 loss enables blood cancer cells to rapidly escape suboptimal BCL-2 inhibition. (A) Western blot confirming CRISPR/Cas9-induced loss of TP-53 expression in independent clones of the RS4;11 human ALL-derived cell line, expressing sgRNAs to target TP53. In all cases, cells were cotreated with an MDM2 inhibitor for 16 hours to induce expression and activation of TP-53, as well as the broad-spectrum caspase inhibitor Q-VD-OpH to prevent late stages of apoptosis that are associated with protein degradation. (B) TP53 wild-type (WT) or TP53-deficient (KO) clones of RS4;11 were treated with 0 to 10 μM venetoclax and their viability determined 24 hours later; the IC50 values are indicated in parentheses. (C) The growth of WT or TP53 KO RS4;11 cells treated continuously with a suboptimal (IC20) dose (supplemental Table 4) of venetoclax (or control treated) was monitored. (D) The outgrowth of TP53 KO RS4;11 cells seeded in a 5 TP53 KO:95 TP53 WT ratio treated continuously with an IC50 dose of venetoclax (or control treated) was monitored by flow cytometric analysis. Data are means ± SD of ≥3 independent experiments. (E) The viability of aliquots of TP53 KO RS4;11 cells or their TP53 WT controls (EV− empty vector) maintained continuously in an IC20 dose of venetoclax for 0, 7, 14, 21, and 28 days (see panel C) treated for 24 hours with 0 to 10 μM venetoclax was determined as in panel B; the IC50 values are indicated in supplemental Table 5. (F) Experiments similar to those with RS4;11 ALL cells (A-B, D) were undertaken with OCI-LY19 cells, a human DLBCL-derived cell line. Data from cell viability (Cell-Titer Glo) or cell competition (fluorescence-activated cell sorting [FACS]) assays represent means ± SD of ≥3 independent experiments.
TP53 loss enables blood cancer cells to rapidly escape suboptimal BCL-2 inhibition. (A) Western blot confirming CRISPR/Cas9-induced loss of TP-53 expression in independent clones of the RS4;11 human ALL-derived cell line, expressing sgRNAs to target TP53. In all cases, cells were cotreated with an MDM2 inhibitor for 16 hours to induce expression and activation of TP-53, as well as the broad-spectrum caspase inhibitor Q-VD-OpH to prevent late stages of apoptosis that are associated with protein degradation. (B) TP53 wild-type (WT) or TP53-deficient (KO) clones of RS4;11 were treated with 0 to 10 μM venetoclax and their viability determined 24 hours later; the IC50 values are indicated in parentheses. (C) The growth of WT or TP53 KO RS4;11 cells treated continuously with a suboptimal (IC20) dose (supplemental Table 4) of venetoclax (or control treated) was monitored. (D) The outgrowth of TP53 KO RS4;11 cells seeded in a 5 TP53 KO:95 TP53 WT ratio treated continuously with an IC50 dose of venetoclax (or control treated) was monitored by flow cytometric analysis. Data are means ± SD of ≥3 independent experiments. (E) The viability of aliquots of TP53 KO RS4;11 cells or their TP53 WT controls (EV− empty vector) maintained continuously in an IC20 dose of venetoclax for 0, 7, 14, 21, and 28 days (see panel C) treated for 24 hours with 0 to 10 μM venetoclax was determined as in panel B; the IC50 values are indicated in supplemental Table 5. (F) Experiments similar to those with RS4;11 ALL cells (A-B, D) were undertaken with OCI-LY19 cells, a human DLBCL-derived cell line. Data from cell viability (Cell-Titer Glo) or cell competition (fluorescence-activated cell sorting [FACS]) assays represent means ± SD of ≥3 independent experiments.
Pharmacologically targeting BCL-2 does not trigger a DNA damage response
In considering how TP-53–deficient leukemic cells out-compete WT cells in response to venetoclax, we initially explored whether venetoclax could indirectly activate TP-53. DNA damage activates TP-53 to drive a transcriptional program promoting apoptosis in hematopoietic cells.5 It has been proposed that venetoclax may cause DNA damage, potentially accounting for impaired apoptosis in TP-53–deficient leukemic cells.18 To avoid the confounding effect of DNA damage resulting from caspase activation in the later stages of apoptosis, we used BAX/BAK-deficient cells known to be resistant to venetoclax-induced cell death.19,20 Agents known to directly cause DNA damage in the absence of BAX/BAK (eg, cisplatin and fludarabine) triggered dose-dependent DNA damage, as revealed by an increase in γH2AX staining. In contrast, no impact on γH2AX staining resulted from inhibition of BCL-2, ruling out the potential for venetoclax to induce direct DNA damage (Figure 4A).
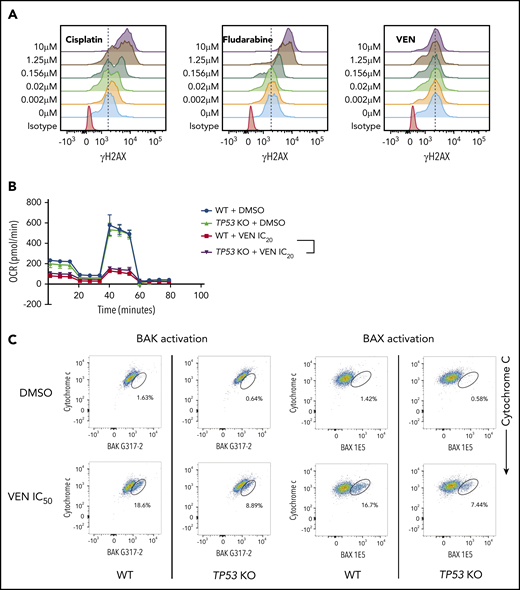
Defects in TP-53 function impair the induction of apoptosis in cancer cells by venetoclax. (A) BAX/BAK double-deficient RS4;11 cells, that are unable to undergo apoptosis, were treated with increasing doses (0-10 μM) of the DNA-damaging drugs cisplatin or fludarabine, or venetoclax (VEN) and γH2AX staining (an indicator of DNA damage) was examined by flow cytometry after 24 hours. Note that cisplatin and fludarabine, but not venetoclax, induced DNA damage, even in the absence of apoptotic cell death. Data are representative of ≥3 independent experiments. (B) WT or TP53 KO clones of MOLM-13 cells were treated with IC20 doses of venetoclax or DMSO (vehicle control) for 3 days. Mitochondrial metabolism was examined by using the Seahorse Mito Stress Test Kit. Error bars indicate SD of 8 replicate samples. (C) Activation of BAX (detected by antibody 1E5) or BAK (detected by antibody G317-2), both early events in apoptosis signaling (x-axes), and loss of cytochrome c (a later event; y-axes) in TP53 WT or TP53 KO RS4;11 cells were determined by flow cytometry 6 hours after treatment with IC50 doses of venetoclax. Data are representative of ≥3 independent experiments.
Defects in TP-53 function impair the induction of apoptosis in cancer cells by venetoclax. (A) BAX/BAK double-deficient RS4;11 cells, that are unable to undergo apoptosis, were treated with increasing doses (0-10 μM) of the DNA-damaging drugs cisplatin or fludarabine, or venetoclax (VEN) and γH2AX staining (an indicator of DNA damage) was examined by flow cytometry after 24 hours. Note that cisplatin and fludarabine, but not venetoclax, induced DNA damage, even in the absence of apoptotic cell death. Data are representative of ≥3 independent experiments. (B) WT or TP53 KO clones of MOLM-13 cells were treated with IC20 doses of venetoclax or DMSO (vehicle control) for 3 days. Mitochondrial metabolism was examined by using the Seahorse Mito Stress Test Kit. Error bars indicate SD of 8 replicate samples. (C) Activation of BAX (detected by antibody 1E5) or BAK (detected by antibody G317-2), both early events in apoptosis signaling (x-axes), and loss of cytochrome c (a later event; y-axes) in TP53 WT or TP53 KO RS4;11 cells were determined by flow cytometry 6 hours after treatment with IC50 doses of venetoclax. Data are representative of ≥3 independent experiments.
Loss of TP-53 does not render mitochondria less susceptible to MOMP
Venetoclax has been reported to enhance metabolic vulnerability by impairing oxidative phosphorylation in AML cells.21 We therefore considered whether TP-53 deficiency could alter mitochondrial metabolism, reducing venetoclax sensitivity.7 We compared real-time metabolic flux in response to venetoclax in wild-type and TP53-deficient cells. Consistent with previous reports, venetoclax substantially impaired the basal oxygen consumption rate, as well as the glycolytic reserve in MOLM-13 (Figure 4B) and MV-4-11 cells (supplemental Figure 3A). TP-53 deficiency, however, neither enhanced the basal respiratory capacity of leukemic cells, nor impaired the reduction in glycolytic capacity induced by venetoclax compared with TP53 wild-type cells (Figure 4B; supplemental Figure 3A).
It has also been postulated that TP-53 deficiency could render leukemic cells less sensitive to induction of apoptosis via MOMP.8 We assessed the sensitivity of mitochondria to MOMP by BH3BIM peptides and found it was unaffected by TP-53 loss (supplemental Figure 3B). Taken together, these findings preclude direct induction of DNA damage or altered mitochondrial function, as explanations for why TP-53 loss confers a competitive advantage to leukemic cells treated for extended periods with venetoclax.
The onset of apoptosis induced by venetoclax is delayed in the absence of TP-53
BH3-mimetics trigger activation of BAX/BAK, leading to MOMP, cytochrome c release, and then caspase activation.22,23 We next investigated whether TP-53 loss could impair the integrity of early (BAX/BAK activation) vs later stages (caspase activation) of apoptosis. After venetoclax exposure, the magnitude of caspase activation was not affected by TP-53 status (supplemental Figure 3C). In contrast, levels of conformationally active BAX and BAK were both distinctly lower in TP53-deficient, compared with wild-type RS4;11 cells after a 6-hour treatment with venetoclax (Figure 4C).5 Of note, total BAX/BAK levels were unaffected by TP-53 loss (supplemental Figure 5C). At 6 hours, TP53-deficient cells also appeared relatively more resistant to venetoclax than TP53-wild-type counterparts. However, these differences diminished over time (16 hours, supplemental Figure 3D; 24 hours, Figure 3B). Overall, we conclude from our mechanistic experiments that deficient TP-53 impairs the earliest stage of apoptosis induction as measured by reduced activation of BAX and BAK, in response to sublethal concentrations of venetoclax.
TP-53 loss also compromises the efficacy of MCL-1 inhibitors in models of human and mouse B-cell leukemia and lymphoma
Having demonstrated that TP-53 loss resulted in gain of fitness for leukemias exposed to venetoclax, we sought to determine whether this observation extended to inhibitors of other prosurvival proteins. We examined BH3-mimetics targeting MCL-1, as clinical trials have recently commenced in hematological malignancies.23-26 To identify factors important for efficacy of MCL-1 inhibitors, a genome-wide CRISPR/Cas9 loss-of-function screening (Figure 5A) was conducted in mouse Eµ-Myc lymphoma-derived cell lines known to be highly MCL-1 dependent.23,27 The top positively selected hits (loss of genes conferring resistance) with the MCL-1 inhibitor S63845 (MCL-1i) were sgRNAs targeting Trp53 and Bax (Figure 5B; supplemental Figure 4A).
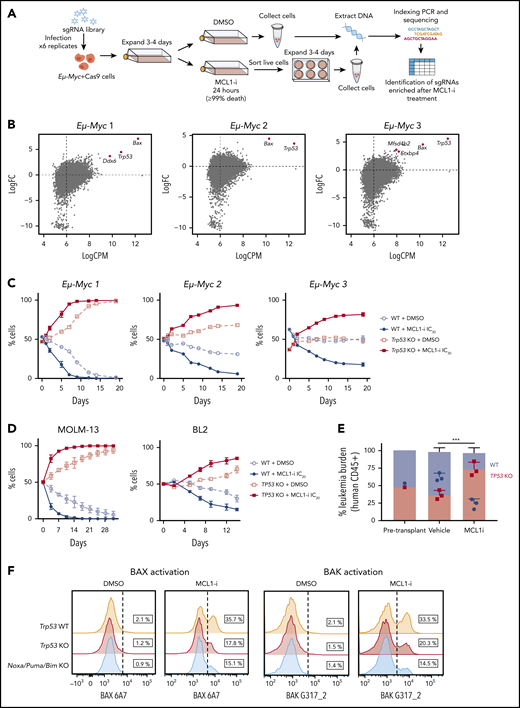
TP53 loss impairs killing induced by an MCL-1 inhibitor. (A) Genome-wide recessive CRISPR screenings to identify genes critical for the action of MCL-1 inhibition. Three independent Cas9-expressing Eµ-Myc mouse lymphoma cell lines were transduced with a library of sgRNAs targeting the mouse genome. These infected cells were expanded and treated with the MCL-1 inhibitor S63845 at doses that killed >99% of cells. The remaining cells that survived were left to expand for 3 to 4 days before DNA was extracted, and targeted amplicon deep sequencing was performed on the MiSeq to identify sgRNAs enriched in cells resistant to MCL-1 inhibitor treatment. (B) Loss of Trp53 and Bax were the top hits for positive selection with the MCL-1i. Enrichment of sgRNAs targeting the indicated genes in 3 independently derived Eµ-Myc lymphoma cell lines treated with the MCL-1i S63845. Eµ-Myc lymphoma cell lines 1-3: AH15A, AF47A, and 560, respectively. (C) The growth of paired Trp53 WT or Trp53 KO Eµ-Myc 1, Eµ-Myc 2, or Eµ-Myc 3 lymphoma cell lines treated continuously with a suboptimal (IC30) dose (supplemental Table 4) of MCL-1i (or control treated) was monitored over 20 days by flow cytometric analysis. See Figure 2C for the experimental outline. (D) Growth competition assay experiments similar to those with mouse Eµ-Myc lymphoma cell lines (panel C) were undertaken with the indicated human cancer-derived cell lines. (E) NSG mice received transplants of an equal number of TP53 WT and TP53 KO MOLM-13 cells. Three days after transplantation, the mice were treated with vehicle or MCL-1i (25 mg/kg once weekly) for 2 weeks. The proportions of TP53 WT (GFP+) and TP53 KO (BFP+) human (CD45+) cells in the peripheral blood were enumerated by flow cytometry. ***P = 2.3 × 10−3 vehicle vs MCL-1i treatment of TP53 KO cells. Data are means ± SD of 5 animals per group from a representative experiment (n = 3 independent experiments). (F) Activation of BAX (detected by antibody 6A7) or BAK (detected by antibody G317-2) in Trp53 WT, Trp53 KO, or Noxa/Puma/Bim KO Eµ-Myc 2 cells were determined by flow cytometry 6 hours after treatment with 150 nM of MCL-1i. Data are representative of ≥3 independent experiments.
TP53 loss impairs killing induced by an MCL-1 inhibitor. (A) Genome-wide recessive CRISPR screenings to identify genes critical for the action of MCL-1 inhibition. Three independent Cas9-expressing Eµ-Myc mouse lymphoma cell lines were transduced with a library of sgRNAs targeting the mouse genome. These infected cells were expanded and treated with the MCL-1 inhibitor S63845 at doses that killed >99% of cells. The remaining cells that survived were left to expand for 3 to 4 days before DNA was extracted, and targeted amplicon deep sequencing was performed on the MiSeq to identify sgRNAs enriched in cells resistant to MCL-1 inhibitor treatment. (B) Loss of Trp53 and Bax were the top hits for positive selection with the MCL-1i. Enrichment of sgRNAs targeting the indicated genes in 3 independently derived Eµ-Myc lymphoma cell lines treated with the MCL-1i S63845. Eµ-Myc lymphoma cell lines 1-3: AH15A, AF47A, and 560, respectively. (C) The growth of paired Trp53 WT or Trp53 KO Eµ-Myc 1, Eµ-Myc 2, or Eµ-Myc 3 lymphoma cell lines treated continuously with a suboptimal (IC30) dose (supplemental Table 4) of MCL-1i (or control treated) was monitored over 20 days by flow cytometric analysis. See Figure 2C for the experimental outline. (D) Growth competition assay experiments similar to those with mouse Eµ-Myc lymphoma cell lines (panel C) were undertaken with the indicated human cancer-derived cell lines. (E) NSG mice received transplants of an equal number of TP53 WT and TP53 KO MOLM-13 cells. Three days after transplantation, the mice were treated with vehicle or MCL-1i (25 mg/kg once weekly) for 2 weeks. The proportions of TP53 WT (GFP+) and TP53 KO (BFP+) human (CD45+) cells in the peripheral blood were enumerated by flow cytometry. ***P = 2.3 × 10−3 vehicle vs MCL-1i treatment of TP53 KO cells. Data are means ± SD of 5 animals per group from a representative experiment (n = 3 independent experiments). (F) Activation of BAX (detected by antibody 6A7) or BAK (detected by antibody G317-2) in Trp53 WT, Trp53 KO, or Noxa/Puma/Bim KO Eµ-Myc 2 cells were determined by flow cytometry 6 hours after treatment with 150 nM of MCL-1i. Data are representative of ≥3 independent experiments.
To validate these hits, we deleted Trp53 in 3 independent Eµ-Myc lymphoma lines (supplemental Figure 4B) and confirmed resistance to MDM2 inhibition (supplemental Figure 4C). Sensitivity to MCL-1i was only minimally affected by Trp53 loss (supplemental Figure 4D). In contrast, deleting Bax rendered the lymphoma cells profoundly resistant to the MCL-1i (>20-fold increase in IC50; supplemental Figure 4E). In long-term cell competition experiments, deletion of Trp53 favored outgrowth in all 3 B-cell lymphoma lines cultured with suboptimal doses of MCL-1i (Figure 5C).
Given these findings, we extended our experiments to human leukemia and lymphoma cell lines. In MOLM-13 cells, TP53 deficiency resulted in an approximate fivefold decrease in sensitivity to an MCL-1i, whereas no change in the IC50 was apparent in BL2 Burkitt lymphoma or RS4;11 cells (supplemental Figure 4F-H). In extended duration competition assays, TP53-deficient MOLM-13 or BL2 cells outcompeted wild-type counterparts when exposed to IC20 MCL-1i drug doses (Figure 5D). This finding was confirmed in vivo after transplantation of MOLM-13 cells into NSG mice and treatment with either MCL-1i or vehicle (Figure 5E). A competitive advantage for TP53-defective variants treated with MCL-1i was further confirmed in MV-4-11 and RS4;11 cell line models in vitro (supplemental Figure 4G-H).
As with venetoclax, there was no evidence that MCL-1 inhibition induced DNA damage in lymphoma cells (supplemental Figure 5A). However, after a 6-hour exposure to a sublethal concentration of MCL-1i, activation of BAX/BAK was impaired in TP53-deficient models of leukemia/lymphoma (Figure 5F; supplemental Figure 5B), akin to our observations with venetoclax.
TP-53 can enhance expression of BH3-only members NOXA and PUMA directly and BIM indirectly, to promote apoptosis.28-31 Deficiency of TP-53 and reduced endogenous baseline BH3 activity may impair the efficacy of exogenously administered BH3-mimetic drugs, particularly at suboptimal doses. To confirm this, we generated Eµ-Myc lymphoma cells that were deficient in Noxa, Puma, and Bim (triple BH3-KO cells) and observed that after a 6-hour treatment with MCL-1i, these cells had impaired BAX/BAK activation, compared with wild-type cells (Figure 5F). Despite similar sensitivities of Noxa/Puma/Bim triple KO, wild-type and Trp53 KO cells to MCL-1i in the 24-hour viability assays (supplemental Figure 6A), a marked competitive advantage was evident for Noxa/Puma/Bim triple-KO cells, compared with either wild-type (supplemental Figure 6B) or Trp53 KO (supplemental Figure 6C) cells in long-term assays cultured in the presence of a sublethal concentration (IC30) of MCL-1i. Of note, a competitive advantage for Noxa/Puma/Bim triple-KO cells was also apparent in control DMSO-treated cultures. This result may be related to MYC-induced predisposition to apoptosis,32 which was reduced by depletion of all 3 BH3-only proteins.31,33
Collectively, these findings suggest that defective TP-53 may increase the threshold for BAX/BAK activation, particularly for sublethal doses of BH3-mimetics administered in single-agent format. We therefore explored the potential for enhancing response to BH3-mimetics by using a combination of drugs to target both BCL-2 and MCL-1 simultaneously, at doses considered individually suboptimal against TP-53 defective leukemia.
Expansion of TP53-deficient populations is not abrogated by cytotoxic drugs, alone or in combination with BH3-mimetics
In clinical practice, venetoclax is approved for use in AML in combination with hypomethylating agents or low-dose cytarabine. Analogous to observations with BH3-mimetics, we noted relative expansion of TP53-deficient MOLM-13 cells in 14-day competition assays, despite continuous exposure to either cytarabine or decitabine (Figure 6A). Furthermore, the competitive advantage of TP53-deleted MOLM-13 cells persisted despite combination of either cytarabine or decitabine with sublethal (IC20) concentrations of BH3-mimetics targeting either BCL-2 or MCL-1 (Figure 6B-C). In 7-day cell competition assays, BCL-2 or MCL-1 inhibitors (at 100 nM), when used as monotherapy, could not restrain outgrowth of TP53-deficient cells (Figure 6D). In contrast, when BCL-2 and MCL-1 inhibitors were combined at one-tenth the dose (10 nM), TP53-deficient and wild-type cells survived in equal proportions for 3 days, with both populations extinguished by day 7 (Figure 6D). These findings suggest that combined targeting of BCL-2 and MCL-1 may counteract the competitive advantage associated with TP53-deficient cells when exposed to BH3-mimetics as single agents at sublethal doses.
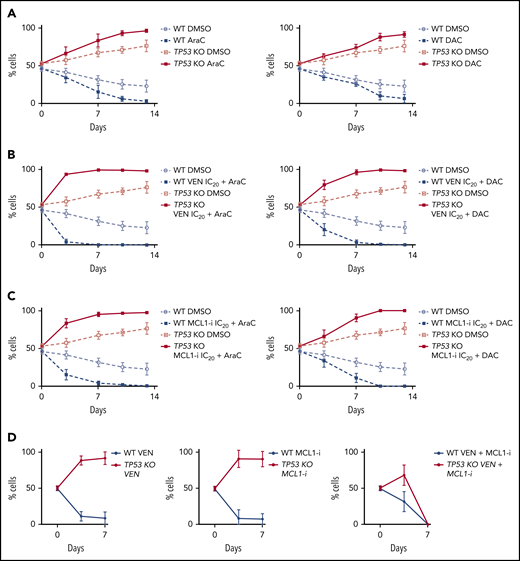
The survival advantage of TP53-deficient cells after BH3-mimetic drug exposure was not suppressed by the combination with either cytarabine or decitabine. (A) The in vitro growth of TP53 WT or TP53 KO MOLM-13 cells was monitored during continuous treatment with a suboptimal dose of cytarabine (100 nM AraC; left) or decitabine (1 µM DAC; right). Data are means ± SD of 4 independent experiments. (B) The growth of TP53 WT or TP53 KO MOLM-13 cells was monitored during continuous treatment with suboptimal doses of cytarabine (100 nM AraC; left panels) or decitabine (1 µM DAC; right panels) in combination with a suboptimal (IC20) dose of venetoclax (100 nM). Data are means ± SD of 3 independent experiments. (C) The growth of TP53 WT or TP53 KO MOLM-13 cells treated continuously with a suboptimal dose of cytarabine (100 nM AraC) or decitabine (1 µM DAC) in combination with a suboptimal (IC20) dose of the MCL-1i (10 nM; or control vehicle treated) was monitored. Data are means ± SD of 3 independent experiments. (D) The in vitro growth of TP53 WT or TP-53 KO MOLM-13 cells was monitored during continuous treatment with venetoclax (100 nM), MCL-1i (100 nM S63845) or combined venetoclax and S63845 (10 nM each drug) over 7 days. Data are means ± SD of 2 independent experiments.
The survival advantage of TP53-deficient cells after BH3-mimetic drug exposure was not suppressed by the combination with either cytarabine or decitabine. (A) The in vitro growth of TP53 WT or TP53 KO MOLM-13 cells was monitored during continuous treatment with a suboptimal dose of cytarabine (100 nM AraC; left) or decitabine (1 µM DAC; right). Data are means ± SD of 4 independent experiments. (B) The growth of TP53 WT or TP53 KO MOLM-13 cells was monitored during continuous treatment with suboptimal doses of cytarabine (100 nM AraC; left panels) or decitabine (1 µM DAC; right panels) in combination with a suboptimal (IC20) dose of venetoclax (100 nM). Data are means ± SD of 3 independent experiments. (C) The growth of TP53 WT or TP53 KO MOLM-13 cells treated continuously with a suboptimal dose of cytarabine (100 nM AraC) or decitabine (1 µM DAC) in combination with a suboptimal (IC20) dose of the MCL-1i (10 nM; or control vehicle treated) was monitored. Data are means ± SD of 3 independent experiments. (D) The in vitro growth of TP53 WT or TP-53 KO MOLM-13 cells was monitored during continuous treatment with venetoclax (100 nM), MCL-1i (100 nM S63845) or combined venetoclax and S63845 (10 nM each drug) over 7 days. Data are means ± SD of 2 independent experiments.
BH3-mimetic combination therapy is highly effective in xenograft models of TP53-defective AML
To model the therapeutic effects of combined BCL-2 and MCL-1 targeting in vivo, NSG mice were engrafted with either MV-4-11 or OCI-AML3 cells comprising an equal ratio of TP53-wild-type and -deficient cells (Figure 7A-B). In the MV-4-11 xenograft model, combined venetoclax/MCL-1i, resulted in significantly longer survival, compared with BH3-mimetics targeting either BCL-2 or MCL-1 alone (Figure 7A). In the OCI-AML3 model, combined BCL-2 and MCL-1 targeting improved survival to a similar extent in the TP-53 WT and KO cohorts, compared with vehicle treatment. This outcome suggests the combination was effective at overcoming the negative impact conferred by loss of TP-53 (Figure 7B).
![BH3-mimetic drug combination therapy is highly effective in xenograft models of TP-53 defective AML. (A) Irradiated NSG mouse recipients of transplants of 105 human MV-4-11 cells harboring the empty vector control (WT TP53) and TP53 KO cells at a 1:1 ratio and followed for Kaplan-Meier survival analysis. Dosing commenced on day 4 after transplant. The mice were divided into treatment groups containing 6 mice each and treated with vehicle, combined venetoclax 75 mg/kg by oral gavage daily (5 days/week for 4 weeks) and S63845 (MCL-1i) 25 mg/kg IV weekly for 4 weeks, or either drug alone (capped line shows treatment interval). Kaplan-Meier (KM) analysis (ethical end points) showed that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle control. *P < .05 compared with vehicle control. (B) Irradiated NSG mice received transplants of 105 human OCI-AML3 cells harboring the empty vector control (WT TP53) or TP53 KO cells and were observed for KM survival analysis. Mice were divided into treatment groups containing 6 to 7 mice in each and dosing was performed as outlined in panel A. KM survival (ethical end points) showing that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle control independent of TP53 status (capped line shows treatment interval). *Significantly enhanced survival (P < .05) compared with vehicle control. (C) Irradiated NRG-SG3 mice received transplants of 106 primary AML cells (TP53-mutant [Y126D] AML; RBWH65). Engraftment was confirmed at week 9 after transplant by detection of hCD45 leukemic cells in peripheral blood. Cohorts of 2 to 4 mice per group were then treated with vehicle (days 1–5), venetoclax 75 mg/kg days 1 to 5 by gavage, S63845 (MCL-1i) 25 mg/kg IV on day 3), or venetoclax 75 mg/kg combined with S63845 25 mg/kg for 10 days and hCD45+ cells in BM enumerated by flow cytometry. (D) Serial BM sampling of NRG-SG3 PDX 01-318-2015 harboring 2 TP53 variants (V173M and V143M). Patient primary sample VAF was 30% for V173M and 27% for V143M. Intrafemoral BM sampling was performed on days 34 and 48 after transplant and when mice were euthanized due to clinical deterioration. The VAF (%) for each TP53 mutation at each time point is shown. Changes in TP53 VAF percentage were quantitated by TP53-targeted NGS and normalized to hCD45+ cells in the BM. (E) Irradiated NRG-SG3 mice received transplants of 106 primary AML cells (harboring 2 TP53 variants, V173M and V143M; 01-328-2015). Engraftment was confirmed at week 2 after transplant by detection of hCD45+ leukemic cells in peripheral blood. Cohorts of 5 mice per group were then treated with vehicle on days 1 to 5, venetoclax 75 mg/kg on days 1 to 5 by gavage, S63845 (MCL-1i) 25 mg/kg IV on day 3, or venetoclax 75 mg/kg combined with S63845 25 mg/kg for 4 weeks. KM survival (ethical end points) showing that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle-treated control mice, independent of TP53 status. *Significantly enhanced survival (P < .05) compared with vehicle control. (F) Activation of BAX (detected by antibody 1E5; x-axes) or loss of cytochrome c; y-axes) in TP53 WT or TP53 KO RS4;11 cells were determined by flow cytometry 6 hours after treatment with IC50 doses of venetoclax or MCL-1i individually, or with venetoclax combined with MCL-1i at IC20 or IC50 doses. Data are representative of ≥3 independent experiments.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020010167/2/m_bloodbld2020010167f7.png?Expires=1763476112&Signature=qgOW7PIU3xldC19gGM0BjmKStA0zEBaRNmkK8tJKe5HlGj-IxNgzTgrOySkSfPKludpErTt8QbwcaOq-IcviFqcL-JuYSsictgefkNu1SyGKd9ng73xc7Ih~7rDhguqTGUbADbDZkSP~0G5VVVOgWPo1ED0fBMvkOWqS6Plg1MP~Dwrtf5hrfSHuNwHS~li0s8MrkMZbpAuOMEQrJrroHs~gwdfP3qU4Lv8wbMRnkz74uxoPZkkpIUuFbBK6My6cBjXl4UcPYRBO2F2bei3nf-8C27mOWykjFmpD2R3NIQ-Ed~ueaX0Gc1ycZ5a2tBP5zw3f5DDWcX6gWzsSOczLxg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
BH3-mimetic drug combination therapy is highly effective in xenograft models of TP-53 defective AML. (A) Irradiated NSG mouse recipients of transplants of 105 human MV-4-11 cells harboring the empty vector control (WT TP53) and TP53 KO cells at a 1:1 ratio and followed for Kaplan-Meier survival analysis. Dosing commenced on day 4 after transplant. The mice were divided into treatment groups containing 6 mice each and treated with vehicle, combined venetoclax 75 mg/kg by oral gavage daily (5 days/week for 4 weeks) and S63845 (MCL-1i) 25 mg/kg IV weekly for 4 weeks, or either drug alone (capped line shows treatment interval). Kaplan-Meier (KM) analysis (ethical end points) showed that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle control. *P < .05 compared with vehicle control. (B) Irradiated NSG mice received transplants of 105 human OCI-AML3 cells harboring the empty vector control (WT TP53) or TP53 KO cells and were observed for KM survival analysis. Mice were divided into treatment groups containing 6 to 7 mice in each and dosing was performed as outlined in panel A. KM survival (ethical end points) showing that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle control independent of TP53 status (capped line shows treatment interval). *Significantly enhanced survival (P < .05) compared with vehicle control. (C) Irradiated NRG-SG3 mice received transplants of 106 primary AML cells (TP53-mutant [Y126D] AML; RBWH65). Engraftment was confirmed at week 9 after transplant by detection of hCD45 leukemic cells in peripheral blood. Cohorts of 2 to 4 mice per group were then treated with vehicle (days 1–5), venetoclax 75 mg/kg days 1 to 5 by gavage, S63845 (MCL-1i) 25 mg/kg IV on day 3), or venetoclax 75 mg/kg combined with S63845 25 mg/kg for 10 days and hCD45+ cells in BM enumerated by flow cytometry. (D) Serial BM sampling of NRG-SG3 PDX 01-318-2015 harboring 2 TP53 variants (V173M and V143M). Patient primary sample VAF was 30% for V173M and 27% for V143M. Intrafemoral BM sampling was performed on days 34 and 48 after transplant and when mice were euthanized due to clinical deterioration. The VAF (%) for each TP53 mutation at each time point is shown. Changes in TP53 VAF percentage were quantitated by TP53-targeted NGS and normalized to hCD45+ cells in the BM. (E) Irradiated NRG-SG3 mice received transplants of 106 primary AML cells (harboring 2 TP53 variants, V173M and V143M; 01-328-2015). Engraftment was confirmed at week 2 after transplant by detection of hCD45+ leukemic cells in peripheral blood. Cohorts of 5 mice per group were then treated with vehicle on days 1 to 5, venetoclax 75 mg/kg on days 1 to 5 by gavage, S63845 (MCL-1i) 25 mg/kg IV on day 3, or venetoclax 75 mg/kg combined with S63845 25 mg/kg for 4 weeks. KM survival (ethical end points) showing that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle-treated control mice, independent of TP53 status. *Significantly enhanced survival (P < .05) compared with vehicle control. (F) Activation of BAX (detected by antibody 1E5; x-axes) or loss of cytochrome c; y-axes) in TP53 WT or TP53 KO RS4;11 cells were determined by flow cytometry 6 hours after treatment with IC50 doses of venetoclax or MCL-1i individually, or with venetoclax combined with MCL-1i at IC20 or IC50 doses. Data are representative of ≥3 independent experiments.
BH3-mimetic drug combination therapy is highly effective in xenograft models of TP-53 defective AML. (A) Irradiated NSG mouse recipients of transplants of 105 human MV-4-11 cells harboring the empty vector control (WT TP53) and TP53 KO cells at a 1:1 ratio and followed for Kaplan-Meier survival analysis. Dosing commenced on day 4 after transplant. The mice were divided into treatment groups containing 6 mice each and treated with vehicle, combined venetoclax 75 mg/kg by oral gavage daily (5 days/week for 4 weeks) and S63845 (MCL-1i) 25 mg/kg IV weekly for 4 weeks, or either drug alone (capped line shows treatment interval). Kaplan-Meier (KM) analysis (ethical end points) showed that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle control. *P < .05 compared with vehicle control. (B) Irradiated NSG mice received transplants of 105 human OCI-AML3 cells harboring the empty vector control (WT TP53) or TP53 KO cells and were observed for KM survival analysis. Mice were divided into treatment groups containing 6 to 7 mice in each and dosing was performed as outlined in panel A. KM survival (ethical end points) showing that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle control independent of TP53 status (capped line shows treatment interval). *Significantly enhanced survival (P < .05) compared with vehicle control. (C) Irradiated NRG-SG3 mice received transplants of 106 primary AML cells (TP53-mutant [Y126D] AML; RBWH65). Engraftment was confirmed at week 9 after transplant by detection of hCD45 leukemic cells in peripheral blood. Cohorts of 2 to 4 mice per group were then treated with vehicle (days 1–5), venetoclax 75 mg/kg days 1 to 5 by gavage, S63845 (MCL-1i) 25 mg/kg IV on day 3), or venetoclax 75 mg/kg combined with S63845 25 mg/kg for 10 days and hCD45+ cells in BM enumerated by flow cytometry. (D) Serial BM sampling of NRG-SG3 PDX 01-318-2015 harboring 2 TP53 variants (V173M and V143M). Patient primary sample VAF was 30% for V173M and 27% for V143M. Intrafemoral BM sampling was performed on days 34 and 48 after transplant and when mice were euthanized due to clinical deterioration. The VAF (%) for each TP53 mutation at each time point is shown. Changes in TP53 VAF percentage were quantitated by TP53-targeted NGS and normalized to hCD45+ cells in the BM. (E) Irradiated NRG-SG3 mice received transplants of 106 primary AML cells (harboring 2 TP53 variants, V173M and V143M; 01-328-2015). Engraftment was confirmed at week 2 after transplant by detection of hCD45+ leukemic cells in peripheral blood. Cohorts of 5 mice per group were then treated with vehicle on days 1 to 5, venetoclax 75 mg/kg on days 1 to 5 by gavage, S63845 (MCL-1i) 25 mg/kg IV on day 3, or venetoclax 75 mg/kg combined with S63845 25 mg/kg for 4 weeks. KM survival (ethical end points) showing that combined treatment with venetoclax/S63845 resulted in significantly longer survival than vehicle-treated control mice, independent of TP53 status. *Significantly enhanced survival (P < .05) compared with vehicle control. (F) Activation of BAX (detected by antibody 1E5; x-axes) or loss of cytochrome c; y-axes) in TP53 WT or TP53 KO RS4;11 cells were determined by flow cytometry 6 hours after treatment with IC50 doses of venetoclax or MCL-1i individually, or with venetoclax combined with MCL-1i at IC20 or IC50 doses. Data are representative of ≥3 independent experiments.
Next, we examined the efficacy of dual BCL-2 and MCL-1 targeting in a PDX mouse model of TP53 Y126D mutant AML (loss of TP-53 function; ClinVar [VCV000265333.7]). After 10 days of therapy, suppression of BM disease burden was highest with combined BH3-mimetic therapy (Figure 7C).
To confirm these observations, a PDX with 2 TP-53 missense variants (TP-53 V173M and V143M; both nonfunctional, ClinVar [VCV000233951.8] and ClinVar [VCV000142657.5]) were treated with vehicle, venetoclax, an MCL-1i or combination BH3-mimetic therapy for 4 weeks. After 2 weeks of treatment, femoral aspirate was harvested from anesthetized mice and TP53 mutation burden assessed by targeted NGS (Figure 7D). The TP53 variant allele frequency (VAF) increased fourfold in cohorts receiving BH3-mimetic monotherapy, suggesting TP53 LOH had evolved for both variants. In contrast, combined venetoclax and MCL-1i therapy suppressed TP53 mutation burden (Figure 7D) and significantly prolonged survival (Figure 7E).
In contrast to BH3-mimetics targeting either BCL-2 or MCL-1 alone, which showed reduced activation of BAX/BAK in TP53 KO RS4;11 cells, a combined BH3-mimetic approach engaging both BCL-2 and MCL-1 simultaneously restored and enhanced early BAX/BAK activation (Figure 7F; supplemental Figure 7). Collectively, these findings show that combined targeting of BCL-2 and MCL-1 overcomes the early apoptotic defect-associated deficient TP-53, facilitating enhanced disease suppression and prolonged survival in diverse models of TP-53–defective AML.
Discussion
BH3-mimetic drugs that directly trigger apoptosis in cancer cells reliant on BCL-2 or its prosurvival relatives have emerged as powerful agents for treating hematologic malignancies.5 After receiving regulatory approvals of venetoclax for CLL and AML, numerous studies34-37 are exploring the clinical impact of venetoclax in other blood cancers. BH3-mimetic drugs targeting MCL-1 have also entered clinical trials for patients with AML, DLBCL, and plasma cell myeloma.24
Clinical experience regarding the impact of TP-53 defects on response to venetoclax in AML and CLL have been paradoxical. In AML, despite a response rate of 55% in older patients with TP53-mutant disease treated with a combination of azacitidine and venetoclax, long-term survival remains poor.38 At relapse, acquisition of a second TP53 mutation or loss of heterozygosity of the wild-type TP53 locus has been observed.11 All these patients received venetoclax in combination with other cytotoxic drugs; therefore, it remains to be determined whether enrichment of TP53 mutation at progression is mediated by resistance to cytotoxic drugs, venetoclax, or both.
In this article, we present a patient who achieved a good clinical response after monotherapy with a BH3-mimetic targeting BCL-2, despite the AML harboring a TP53 P151H mutation and monosomy 17 (Figure 1A). Furthermore, the TP53-mutation burden also diminished with therapy. Although we cannot exclude the possibility that a coexisting IDH1 mutation conferred BCL-2 dependency, akin to that seen with mutant IDH2,39 this case illustrated that TP-53 deficiency does not preclude responses to BCL-2–targeted therapy in AML. In contrast, examination of 4 other patients without a reduction in marrow blasts revealed interval expansion of TP53-mutant clones upon exposure to venetoclax relative to TP53 wild-type populations. This suggests that, in some cases, defective TP-53 may confer resistance to BCL-2–targeted therapy.
TP53 loss as a cause of venetoclax resistance was previously identified from a genome-wide CRISPR screen in AML.7,8 Using a similar approach, we show for the first time an association between TP53 loss and resistance to MCL-1 inhibitors, indicating a potential class effect in relation to TP53 integrity and BH3-mimetic activity. These findings are unexpected, as the prosurvival targets of BH3-mimetics lie downstream of TP-53.
Using engineered TP-53–deficient leukemic models, we have made the surprising finding that TP-53 loss does not impair short-term killing upon exposure to either BCL-2 or MCL-1 inhibitors. Instead, we show a more subtle phenotype, with TP-53 deficiency only revealing a competitive advantage relative to wild-type counterparts after extended exposure to sublethal BH3-mimetics targeting either BCL-2 or MCL-1. In contrast, Bax deletion, also identified from the MCL-1i CRISPR screening, rendered cells profoundly resistant to BH3-mimetics (supplemental Figure 4E).
Mechanistically, the critical question is how TP-53 deficiency impairs the activity of BH3-mimetics which are thought to have their function downstream. We found no evidence that venetoclax activated upstream DNA damage signals sensed by TP-53. There was also no impact of TP-53 deficiency on basal mitochondrial respiration or venetoclax-induced suppression of oxidative phosporylation.40,41 In contrast, we make the unexpected observation that TP-53 deficiency impairs the ability of a suboptimal dose of BH3-mimetics targeting either BCL-2 or MCL-1 to activate BAX and BAK, critical effectors of apoptosis. TP-53 can directly or indirectly induce the expression of several BH3-only proteins (NOXA, PUMA, and BIM)28-31 Upon administration of venetoclax, endogenous BH3-only proteins already bound to BCL-2 may be released, allowing for activation of BAX and BAK. In the setting of TP-53 deficiency, we propose that the amount of endogenous BH3-only proteins that are released from BCL-2 by a sublethal dose of venetoclax is reduced, impairing efficiency of BAX or BAK activation by a suboptimal dose of venetoclax and raising the early apoptotic threshold.
To highlight the importance of endogenous BH3-only proteins regulated by TP-53, we showed that lymphoma cells triple-deficient in Noxa, Puma, and Bim also displayed impaired early activation of BAX and BAK in response to suboptimal BH3-mimetic doses, resulting in a competitive advantage in long-term assays over WT cells. Interestingly, triple BH3-only KO cells also outcompeted TP-53-deficient cells, perhaps because expression of BIM and even PUMA can be regulated via TP-53-independent processes.31,42 Impaired BAX/BAK activation in TP-53 KO cells was remedied by combining sublethal doses of BH3-mimetics so that BCL-2 and MCL-1 could be targeted simultaneously. This strategy led to enhanced short-term activation of BAX/BAK and greater long-term control of TP-53-deficient AML. In contrast, BH3-mimetics combined with cytotoxic drugs failed to suppress the long-term competitive advantage conferred by TP-53–deficient cells.43
Our results have several important clinical implications. First, prolonged exposure of TP-53-deficient malignant cells to a sublethal dose of a BH3-mimetic targeting either BCL-2 or MCL-1 should be avoided. Second, our results suggest that targeting BCL-2 and MCL-1 simultaneously with BH3-mimetics in TP53 mutant AML may be more effective than either drug in combination with conventional cytotoxic or hypomethylating agents. Third, if the activity of MCL-1 inhibitors in TP-53–deficient AML can be enhanced through combination with venetoclax, the need to maximize the dose of MCL-1 inhibitors to obtain a clinical response should be lessened, thereby widening the therapeutic window for this emerging drug class. Last, our findings illustrate the dual importance of TP-53 as a facilitator of optimal proapoptotic drug activity, not only via its role as a sensor of upstream cell stress (eg, DNA damage), but also via its role in regulating the earliest stages of apoptosis induction by BH3-mimetics targeting downstream prosurvival BCL-2 family members.
Our work provides a rationale for exploring BH3-mimetics in combination as an approach to subverting the competitive advantage associated with TP-53–defective leukemic cells and to enhancing both short, and also long-term outcomes in patients with TP53 mutant disease. Indeed, several phase 1b trials combining inhibitors of MCL-1 and BCL-2 in patients with AML are in progress, suggesting that this dual BH3-mimetic targeting approach may be clinically feasible (registered on www.clinicaltrials.gov as #NCT03672695, #NCT03797261).
Processed sequencing data will be made available, subject to a data-transfer agreement. The data will be restricted to ethically approved research into blood cell malignancies and cannot be used to assess germline variants.
The exome sequencing case is stored at the European Genome Archive (accession number EGAS00001004841).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who enrolled in the venetoclax clinical trials; S. Cory for helpful suggestions; M. Herold and A. Kueh for assistance with CRISPR/Cas9 screening; and S. Monard, N. Sprigg, S. Wilcox, C. White, and the Australasian Leukaemia and Lymphoma Group (ALLG) tissue bank for technical assistance and provision of vital samples and reagents. The visual abstract was created using BioRender.com.
This work was supported by fellowships and grants from the Australian National Health and Medical Research Council (NHMRC; Program Grants 1016701 [R.M.K., A.S., and D.C.S.H.] and 1113577 [A.W.R.]; Research Fellowships 1139607 [A.K.], 1140851 [D.A.S.], 1020363 [A.S.], 1079560 [A.W.R.], and 1156024 [D.C.S.H.]; Investigator Grants 1176175 [F.C.B.], 1174902 [A.W.R.], and 1157263 [C.B.]; Project Grants 1140906 [D.A.S.], 1162809 [A.H.W.], and 1086291 [G.L.K.]; and Ideas Grants 2002618 and 2001201 [G.L.K.]); the Leukemia and Lymphoma Society of America (Fellowship 5467-18 [R.T.]; Specialized Center of Research [SCOR] grant 7015-18 (R.M.K., A.S., A.W.R., A.H.W., G.L.K., and D.C.S.H.); a Cure Cancer and Cancer Australia grant (1186003 [R.T.]); Victorian Cancer Agency (Mid-career Research Fellowship [MCRF] 17028 [G.L.K.], MCRF Fellowship 19011 [D.M.M.], and grant 15018 [I.J.M., A.W.R., and A.H.W.); Cancer Council Victoria grants-in-aid 1086157 and 1147328 (G.L.K.), 1124178 (I.J.M.), and 1141740 (D.M.M.); Leukaemia Foundation of Australia (the Bill Long Charitable Trust PhD Clinical Scholarship [E.C., A.S., and G.L.K.]); fellowships from Novartis Foundation for Medical-Biological Research and the Swiss National Science Foundation (P400PM-180807 [S.S.G.]); the Felton Bequest (I.J.M.); the estate of Anthony (Toni) Redstone OAM (A.S. and G.L.K.); the Craig Perkins Cancer Research Foundation (G.L.K.); the Dyson Bequest (G.L.K.); Medical Research Future Fund grant 1141460 (A.H.W.); Tour de Cure Foundation (RSP-212-2020); and the Australian Cancer Research Foundation. This work was made possible through Victorian State Government Operational Infrastructure Support (OIS) and Australian Government NHMRC Independent Research Institute Infrastructure Support (IRIIS) Scheme.
Authorship
Contribution: R.T., S.T.D., D.M., F.C.B., A.S., A.W.R., D.C.S.H., G.L.K., and A.H.W. designed the research; R.T., S.T.D., D.M., E.C., F.C.B., M.X.S., M.A.D., V.L., S.M., N.S.A., B.R., S.S.G., T.M.D., C.D.R., C.C., Z.X., T.M., G.P., C.B., and L.T. performed the research; C.B., S.W.L., B.J.A., R.M.K., A.S., M.S., S.B., A.K., D.A.S., and A.B. contributed vital new reagents or analytical tools; C.F., M.C., E.C., R.T., S.T.D., S.S.G., D.M., F.C.B., I.J.M., R.M.K., A.S., A.W.R., D.C.S.H., G.L.K., and A.H.W. analyzed and interpreted data; R.T., S.T.D., D.M., F.C.B., A.S., A.W.R., D.C.S.H., G.L.K., and A.H.W. performed the statistical analysis; R.T., S.T.D., D.M., A.S., A.W.R., D.C.S.H., F.C.B., G.L.K., and A.H.W. wrote the manuscript; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: M.S. and S.B. are employees of Servier. R.T., S.T.D., E.C., C.F., M.X.S., M.A.D., T.M.D., C.D.R., C.C., Z.X., R.M.K., I.J.M., A.S., A.W.R., D.C.S.H., and G.L.K. are employees, and N.A. and A.H.W. are former employees, of the Walter and Eliza Hall Institute, which receives milestone and royalty payments related to venetoclax. A.H.W. and A.W.R. have received research funding from Servier and Abbvie. D.C.S.H. has received research funding from Genentech. A.S., A.W.R., D.C.S.H., G.L.K., and A.H.W. have received research funding from Servier. The remaining authors declare no competing financial interests.
The current affiliation for B.R. is the Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Melbourne, VIC, Australia.
The current affiliation for B.J.A. is the Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA.
Correspondence: Andrew Wei, Australian Centre for Blood Diseases, Monash University, 99 Commercial Rd, Melbourne, VIC 3004, Australia; e-mail: andrew.wei@monash.edu; Gemma Kelly, Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Melbourne, VIC 3052, Australia; e-mail: gkelly@wehi.edu.au; Fiona Brown, Australian Centre for Blood Diseases, Monash University, 99 Commercial Rd, Melbourne, VIC 3004, Australia; e-mail: fiona.brown2@monash.edu; and David Huang, Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Melbourne, VIC 3052, Australia; e-mail: huang_d@wehi.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal