

Key Points
Tfpi−/− embryos have severe vascular pathology with associated cellular death in the CNS, but not in other organs.
Removing FV from Tfpi−/− embryos completely ameliorates the CNS pathology.

Abstract
Tissue factor pathway inhibitor (TFPI) inhibits proteases in the blood coagulation cascade that lead to the production of thrombin, including prothrombinase (factor Xa [FXa]/FVa), the catalytic complex that directly generates thrombin. Thus, TFPI and FV are directly linked in regulating the procoagulant response. Studies using knockout mice indicate that TFPI and FV are necessary for embryogenesis, but their contributions to vascular development are unclear. We performed extensive histological analyses of Tfpi−/− and Tfpi−/−F5−/− mouse embryos to investigate the importance of the interplay between TFPI and FV in regulating hemostasis and vascular development during embryogenesis. We observed normal tissue development throughout Tfpi−/− embryos, except in the central nervous system (CNS). The CNS displayed stunted brain growth, delayed development of the meninges, and severe vascular pathology characterized by the formation of glomeruloid bodies surrounding areas of cellular death, fibrin deposition, and hemorrhage. Removing FV from Tfpi−/− embryos completely ameliorated their brain pathology, suggesting that TFPI dampens FV-dependent procoagulant activity in a manner that modulates cerebrovascular development. Thus, we have identified a previously unrecognized role for TFPI activity within the CNS. This TFPI activity likely diminishes an effect of excess thrombin activity on signaling pathways that control cerebral vascular development.
Introduction
Tissue factor pathway inhibitor (TFPI) is a multivalent Kunitz-type serine protease inhibitor that directly inhibits factor Xa (FXa) and provides FXa-dependent feedback inhibition of the tissue factor (TF)–FVIIa catalytic complex, which initiates coagulation through activation of FIX and FX.1-4 The inhibitory activity of TFPI is mediated by simultaneous inhibition of TF-FVIIa and FXa in the TF-FVIIa-FXa ternary complex.5 By inhibiting these blood coagulation proteases, TFPI also controls intracellular signaling events mediated by protease-activated receptor (PARs) that modulate vascular development,6 cellular migration,7,8 and tumor angiogenesis.9
The importance of TFPI in development is illustrated by the embryonic death observed in homozygous mice lacking the first Kunitz (K1) domain of TFPI (Tfpitm1Gjb; Tfpi−/−).10 On a mixed 129-C57BL/6 genetic background, ∼60% of Tfpi−/− embryos die during midgestation between embryonic day (E)9.5 and E11.5. The remaining embryos die later in gestation, between E12.5 and birth. Midgestational embryonic death is associated with apparent vascular abnormalities of the yolk sac with associated hemorrhage. The later-stage embryos had normal vasculature but developed hepatic fibrin deposition suggestive of a consumptive coagulopathy. Rare intravascular thrombi were also observed in the brain of some embryos, while massive hemorrhage with significant loss of brain matter was observed in others.10,11 However, the origin of these brain phenotypes remains unclear.
TFPI also inhibits the FVa-FXa catalytic complex (prothrombinase),12 which converts prothrombin to thrombin. Thrombin is the terminal blood coagulation protease that produces a fibrin-rich blood clot. Thrombin also is a potent activator of PARs on platelets and endothelium.13 Prothrombinase inhibition is mediated by multiple interactions between TFPI and prothrombinase that directly inhibit the ability of prothrombinase to convert prothrombin to thrombin,12,14 as well as by TFPI sterically constraining thrombin from fully cleaving the FV B-domain.15 Thus, TFPI and FV are directly linked in the biological regulation of thrombin production and availability for PAR activation. For example, PAR4 (F2rl3) is the major thrombin-activated PAR on mouse platelets.16 Its absence partially rescues Tfpi−/− mice from embryonic lethality, implicating TFPI as a physiologically relevant inhibitor of thrombin-dependent platelet procoagulant activity during murine embryogenesis.17
Homozygosity for FV deletion (F5−/−) also results in embryonic lethality in mice.18 Deranged vascular development is present in F5−/− yolk sacs contributing to midgestational death. Similar to Tfpi−/− embryos, a portion of F5−/− embryos survive this midgestation bottleneck, developing normally until succumbing perinatally from severe intra-abdominal hemorrhage induced by birthing trauma. However, the global vasculature in these embryos appears otherwise intact.18 Thus, FV is necessary for surviving the hemostatic challenges associated with birth, but the contribution of FV to embryonic vascular development is not fully understood.
In addition to the F5−/− mouse, several F5 transgenic mouse models have been developed.19,20 Combined with the Tfpi−/− mouse, these models provide an opportunity to further examine contributions of the blood coagulation system to hemostasis and vascular development during embryogenesis. Here, we have performed extensive, detailed histological analyses of multiple tissues from Tfpi−/− embryos and identified a role for TFPI-dependent inhibition of FV-driven coagulation necessary for proper cerebral vascular development.
Materials and methods
Generation of mice and embryos
Mice heterozygous for the TFPI-K1 null allele (Tfpitm1Gjb;Tfpi+/−)10 were a generous gift from George Broze, Jr. (Washington University, St Louis, MO). Tfpi+/− mice were backcrossed to C57BL/6J (stock #000664, The Jackson Laboratory) for >10 generations and maintained as an independent line within our mouse colony. Timed matings were performed by crossing Tfpi+/− male and female mice. Mice with a completely FV null allele (F5tm1Dgi stock #004078, The Jackson Laboratory, F5−) and transgenic mice with bacterial artificial chromosomes expressing FV under control of either the murine hepatocyte-specific albumin promoter FV (B6;SJL-Tg[Alb-F5]2Dgi/J stock #007244, The Jackson Laboratory, F5LiverTg) or the murine platelet factor 4 promoter (B6;SJL-Tg[Cxcl4-F5]1Dgi/J stock #007243, The Jackson Laboratory, F5PltTg) have been previously described.20 Genotyping for Tfpi−, F5−, F5LiverTg, and F5PltTg were performed by polymerase chain reaction analysis of tail DNA using primers as described previously.10,20 Crossbreeding experiments using F5−/− mice carrying liver- or platelet-specific transgenes were performed to create F5−/−F5LiverTg and F5−/−F5PltTg mice. F5−/− mice are viable when they co-inherit the F5LiverTg or F5PltTg.20 F5−/−F5LiverTg and F5−/−F5PltTg were separately bred onto the Tfpi-deficient mouse background to produce F5−/−F5LiverTg and F5−/−F5PltTg embryos that are also Tfpi−/−.
Tissue retrieval
Pregnant females were anesthetized using isoflurane inhalant. The uterus containing the embryos was removed. The mother was immediately killed by anesthetic overdose and cervical dislocation. The embryos were retrieved and processed according to National Institutes of Health Guidelines for Euthanasia for Rodent Fetuses and Neonates (https://oacu.oir.nih.gov/sites/default/files/uploads/arac-guidelines/b4_rodent_euthanasia_pup.pdf). Each embryo was separated from the uterus and placenta, washed in 4°C phosphate-buffered saline, and placed in 4°C 10% formaldehyde buffered with phosphate-buffered saline. After 7 days, the embryo midline was sagittally transected and each half processed for histology. In some embryos, brains were coronally sectioned for histological evaluation. The gestational age of embryos was determined by measuring crown to rump length, Theiler’s stage criteria,21 and histological examination of lung, kidney, and brain.
Histology
Paraffin-embedded tissue was cut in 5-µm sections and visualized using hematoxylin and eosin (H&E), trichrome, or Martius Scarlet blue (MSB) staining. Primary immunohistochemistry antibodies included anti-laminin-1 (L9393; Sigma-Aldrich, St. Louis, MO) and anti-fibrinogen (YNGMFBG7S; Accurate Chemical & Scientific Corp, Westbury, NY). Species specific immunoglobulin G comparable to the test antibodies served as controls. Secondary labeling used species specific ImmPRESS (horseradish peroxidase) polymers (MP-7405 and MP-7401; Vector Laboratories, Burlingame, CA) followed by detection with NovaRed (#SK-4805, Vector Laboratories) and counterstaining with hematoxylin (H-3401, Vector Laboratories). Immunofluorescent staining was performed using anti-laminin-1 (L9393, Sigma-Aldrich), antithrombomodulin (AF-3894; R&D Systems, Minneapolis, MN), anti-platelet-derived growth factor receptor β (anti-PDGFR-β; #LS-C88709; LifeSpan Biosciences, Seattle, WA), anti-microglial ionized calcium-binding adapter molecule 1 (019-19741; Waco, Richmond, VA), and a polyclonal rabbit antibody to the extracellular domain of mouse G protein coupled receptor 124 (Gpr124, a gift from Calvin Kuo, Stanford University, Stanford, CA). Secondary antibodies with a fluorescent conjugate at wavelength 568, 594, or 647 nm were from Jackson ImmunoResearch Labs (West Grove, PA). Species-specific immunoglobulin G comparable to the test antibodies served as controls. A Motic Easy scan imaging system (Motic, Hong Kong) scanned immunohistochemistry slides. A Nikon Eclipse Ti2 inverted microscope with a DS-Ri2 high-speed color camera using a low-magnification 40×/0.95 or a high-magnification 100× oil/1.45 numerical aperture oil objective (Nikon Instruments, Melville, NY) acquired immunofluorescent images. Images were analyzed with the Nikon NIS-Elements software platform and processed with Imaris multichannel microscopy software (Bitplane, Concord, MA). Image formatting was performed in Adobe Photoshop CS-6 and Adobe Illustrator CS-6. (Adobe, San Jose, CA).
Study approval
The institutional animal care and use committees of the Medical College of Wisconsin and University of Michigan approved all animal experiments.
Results
Tfpi−/− embryos exhibited stunted brain development with cerebrovascular anomalies, hemorrhage, and cellular death
Microscopic examination of the brains from E15.5 Tfpi+/+ embryos stained with H&E revealed age-appropriate brain development. We observed normal brain architecture that filled the calvarium and exhibited normal growth of the choroid plexus. In contrast, embryonic brains from Tfpi−/− littermates were smaller and did not fill the calvarium. The choroid plexus was underdeveloped, and there were numerous enlarged, misshapen blood vessels throughout the brain parenchyma (Figure 1A; supplemental Figure 1A, available on the Blood Web site). Examination of sections stained with trichrome or MSB identified the enlarged vessels observed with H&E stain to be well-demarcated, multifocal vascular structures resembling renal glomeruli known as glomeruloid bodies (Figure 1B). Glomeruloid bodies are a diagnostic feature of the brain cancer, glioblastoma multiforme.22,23 In contrast to the architecture of glomeruloid bodies, vasculature of Tfpi+/+ embryos had parallel walls lined with endothelial cells and often contained red blood cells (RBCs) (supplemental Figure 1B). In Tfpi−/− embryos, large areas with hemorrhage and cellular death were most readily observed in MSB-stained tissue. These areas were encircled with glomeruloid bodies and appeared to coincide with their formation (Figure 1C). Glomeruloid bodies were present in the brains of 12 of 12 Tfpi−/− embryos obtained from 7 litters between E12.5 and E16.5 but were not present in any of 50 Tfpi+/+ or Tfpi+/− embryos, indicating that deletion of the K1 domain of TFPI results in complete penetrance of this cerebrovascular phenotype in homozygous carriers.
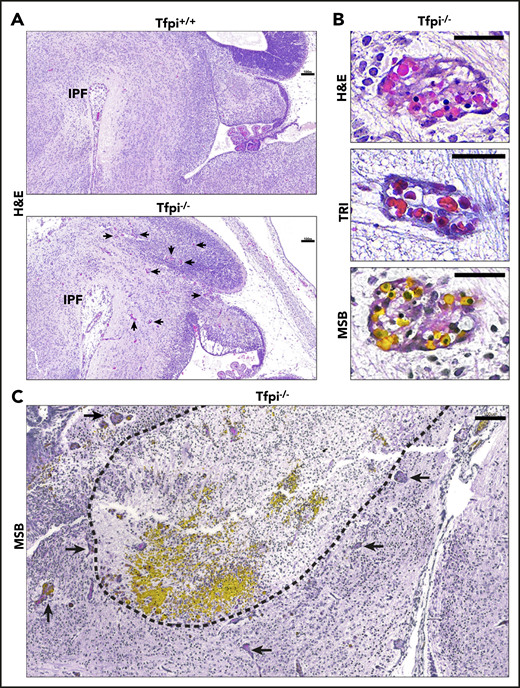
E15.5 sagittal sections of Tfpi−/−brains at the level of the interpeduncular fossa displayed large areas of cellular death and associated hemorrhage surrounded by vascular anomalies called glomeruloid bodies. (A) H&E staining shows that the Tfpi+/+ brain fills the entire cavity of the skull, while the Tfpi−/− brain is smaller and underdeveloped, with a large space between the brain and the calvarium. Arrows identify enlarged congested blood vessels in the Tfpi−/− brain (scale bars, 100 µm). IPF, interpeduncular fossa. (B) High magnification of the enlarged congested vessels in the Tfpi−/− brain identified them as glomeruloid bodies (scale bars, 30 µm). H&E: a multicompartmental vessel with pink amorphous material suggestive of fibrin clot and lumens containing nucleated RBCs. Trichrome (TRI): a multicompartmental vessel with septa containing collagen and nucleated RBCs within the lumens. MSB: a multicompartmental vessel containing numerous yellow nucleated RBCs and pink to red fibrin lining the lumens. (C) MSB staining highlighted the brain pathology of Tfpi−/− embryos. The dotted black line encircles a large area of cell death (pyknotic nuclei and loss of parenchyma) and hemorrhage (pools of yellow-stained nucleated RBCs). Arrows identify numerous glomeruloid bodies surrounding the area of necrosis and hemorrhage (scale bar, 100 µm).
E15.5 sagittal sections of Tfpi−/−brains at the level of the interpeduncular fossa displayed large areas of cellular death and associated hemorrhage surrounded by vascular anomalies called glomeruloid bodies. (A) H&E staining shows that the Tfpi+/+ brain fills the entire cavity of the skull, while the Tfpi−/− brain is smaller and underdeveloped, with a large space between the brain and the calvarium. Arrows identify enlarged congested blood vessels in the Tfpi−/− brain (scale bars, 100 µm). IPF, interpeduncular fossa. (B) High magnification of the enlarged congested vessels in the Tfpi−/− brain identified them as glomeruloid bodies (scale bars, 30 µm). H&E: a multicompartmental vessel with pink amorphous material suggestive of fibrin clot and lumens containing nucleated RBCs. Trichrome (TRI): a multicompartmental vessel with septa containing collagen and nucleated RBCs within the lumens. MSB: a multicompartmental vessel containing numerous yellow nucleated RBCs and pink to red fibrin lining the lumens. (C) MSB staining highlighted the brain pathology of Tfpi−/− embryos. The dotted black line encircles a large area of cell death (pyknotic nuclei and loss of parenchyma) and hemorrhage (pools of yellow-stained nucleated RBCs). Arrows identify numerous glomeruloid bodies surrounding the area of necrosis and hemorrhage (scale bar, 100 µm).
Glomeruloid bodies in Tfpi−/− brain had fibrin(ogen) deposition and endothelial cell hyperplasia
The MSB-stained brain tissue from the Tfpi−/− embryos had red to pink areas, suggesting fibrin deposition (Figure 1B). This was confirmed by immunohistochemical studies identifying fibrin(ogen) inside the glomeruloid bodies, within the brain parenchyma surrounding the glomeruloid bodies, and within areas of cellular death (Figure 2A). Immunohistochemistry for laminin, a vascular basement membrane protein,24 clearly delineated the glomeruloid bodies within brain parenchyma (Figure 2B). Immunofluorescent studies costaining for laminin and the endothelial marker thrombomodulin were used to further examine brain vasculature (Figure 2C). The Tfpi+/+ cerebral vasculature was well organized, with endothelial cells lining the vessel lumen surrounded by a well-organized laminin-stained basement membrane. Laminin staining of the Tfpi−/− brain highlighted the enlarged multilobular and thin-walled structure of the glomeruloid bodies, while thrombomodulin staining was disorganized, indicating endothelial cell disarray. These characteristics are similar to those described for glomeruloid bodies present in glioblastoma multiforme, which are characterized by aggregates of focally anastomosing capillaries with narrow lumina and endothelial cell hyperplasia.23
![Glomeruloid bodies had fibrin deposition and endothelial cell disarray. (A) Sagittal sections of E15.5 littermate brains stained with anti-fibrin(ogen). The inset of the Tfpi−/− brain shows fibrin(ogen) deposits inside and outside the vascular compartment, suggesting vasculature leakage around the glomeruloid bodies (arrows). Areas of cell death characterized by pyknotic nuclei were adjacent to the glomeruloid bodies (arrowheads). Fibrin(ogen) deposition was not observed in the Tfpi+/+ brain (scale bars, 100 µm [inset, 30 µm]). (B) Sagittal sections of E15.5 littermate brains stained with an anti-laminin antibody. Numerous glomeruloid bodies throughout multiple areas of the Tfpi−/− brain were outlined by laminin staining of the basement membrane. Glomeruloid bodies were not present in Tfpi+/+ brain (scale bars, 100 µm [inset, 30 µm]). (C) Immunofluorescence of littermate brains stained for laminin (red/yellow) and thrombomodulin (THBD; green), with 4′,6-diamidino-2-phenylindole (DAPI)–stained nuclei (blue). Tfpi+/+ brain had well-organized vasculature with thrombomodulin-stained endothelial cells internal to the laminin-stained vascular basement membrane. Tfpi−/− embryo brain had disorganized multilobular vasculature containing thrombomodulin-stained endothelial cells in disarray surrounded by a laminin-stained basement membrane (scale bars, 10 µm).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/2/10.1182_blood.2020006054/3/m_bloodbld2020006054f2.png?Expires=1769277043&Signature=3rsDVQqG09kRYipbEaRWe2HolfwQQjoNqekDaJ869s4UPe~yQSpT-9vkFKvg9m2ytLKG9hZN5kInYyw3fTBgG61vVsS0MfHqeOi5a~F0gi7SJcRzHZ5pv97FLrcvlI0d01bQPsDOSKypS1zr8K0-TV77csrJx-oWey6NKAmFGqqxiT4xorkStfKYMcSjsVgOPda9wEaIVjEZO9m18SealC7ywYZoIzGEx-KqRwAORBvsOh96cl8lwe3BhIYBoZSvtcj4oof5qRhgFgvSgq6lsYmRKJji9Foybqrllm1W6fK0dqQ-ET4p13mTCKZp9StOi-weQp1bldf0Q631gf7wuw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Glomeruloid bodies had fibrin deposition and endothelial cell disarray. (A) Sagittal sections of E15.5 littermate brains stained with anti-fibrin(ogen). The inset of the Tfpi−/− brain shows fibrin(ogen) deposits inside and outside the vascular compartment, suggesting vasculature leakage around the glomeruloid bodies (arrows). Areas of cell death characterized by pyknotic nuclei were adjacent to the glomeruloid bodies (arrowheads). Fibrin(ogen) deposition was not observed in the Tfpi+/+ brain (scale bars, 100 µm [inset, 30 µm]). (B) Sagittal sections of E15.5 littermate brains stained with an anti-laminin antibody. Numerous glomeruloid bodies throughout multiple areas of the Tfpi−/− brain were outlined by laminin staining of the basement membrane. Glomeruloid bodies were not present in Tfpi+/+ brain (scale bars, 100 µm [inset, 30 µm]). (C) Immunofluorescence of littermate brains stained for laminin (red/yellow) and thrombomodulin (THBD; green), with 4′,6-diamidino-2-phenylindole (DAPI)–stained nuclei (blue). Tfpi+/+ brain had well-organized vasculature with thrombomodulin-stained endothelial cells internal to the laminin-stained vascular basement membrane. Tfpi−/− embryo brain had disorganized multilobular vasculature containing thrombomodulin-stained endothelial cells in disarray surrounded by a laminin-stained basement membrane (scale bars, 10 µm).
Glomeruloid bodies had fibrin deposition and endothelial cell disarray. (A) Sagittal sections of E15.5 littermate brains stained with anti-fibrin(ogen). The inset of the Tfpi−/− brain shows fibrin(ogen) deposits inside and outside the vascular compartment, suggesting vasculature leakage around the glomeruloid bodies (arrows). Areas of cell death characterized by pyknotic nuclei were adjacent to the glomeruloid bodies (arrowheads). Fibrin(ogen) deposition was not observed in the Tfpi+/+ brain (scale bars, 100 µm [inset, 30 µm]). (B) Sagittal sections of E15.5 littermate brains stained with an anti-laminin antibody. Numerous glomeruloid bodies throughout multiple areas of the Tfpi−/− brain were outlined by laminin staining of the basement membrane. Glomeruloid bodies were not present in Tfpi+/+ brain (scale bars, 100 µm [inset, 30 µm]). (C) Immunofluorescence of littermate brains stained for laminin (red/yellow) and thrombomodulin (THBD; green), with 4′,6-diamidino-2-phenylindole (DAPI)–stained nuclei (blue). Tfpi+/+ brain had well-organized vasculature with thrombomodulin-stained endothelial cells internal to the laminin-stained vascular basement membrane. Tfpi−/− embryo brain had disorganized multilobular vasculature containing thrombomodulin-stained endothelial cells in disarray surrounded by a laminin-stained basement membrane (scale bars, 10 µm).
Tfpi−/− embryos had glomeruloid bodies in multiple areas of the brain and spinal cord, but not in other vascular beds
Examination of sagittal sections of Tfpi−/− embryonic brain revealed extensive pathology in the midbrain (Figure 2B). Coronal sectioning of the brain was then performed to further define affected regions of the brain (Figure 3) with contrasting coronal sections of Tfpi+/+ embryo brains illustrating normal vascular development (supplemental Figure 2). These studies found that Tfpi−/− embryos also developed glomeruloid bodies within the forebrain and hindbrain, indicating that multiple areas of the brain were affected by this altered vascularization process (Figure 3A). Coronal sections of the Tfpi−/− embryonic brain also identified altered vasculature along the pial surface, within the subventricular zone, and within the lateral ganglionic eminence of the forebrain (Figure 3B). Glomeruloid bodies also were present within the spinal cord (Figure 3B). In contrast, other tissues had normal growth, architecture, and vasculature (supplemental Figure 3). Thus, the formation of glomeruloid bodies in Tfpi−/− embryos was limited to the embryonic central nervous system (CNS), indicating a tissue-specific etiology for these defects.
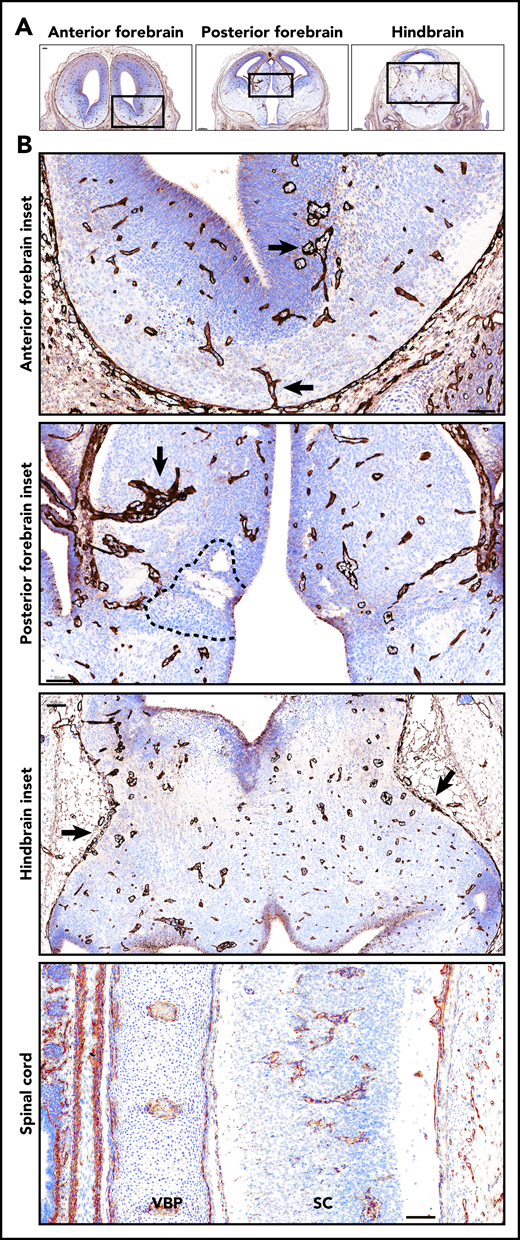
Glomeruloid bodies were present in the forebrain, hindbrain, and spinal cord of E14 Tfpi−/−brain. (A) Coronal sections of anterior forebrain, posterior forebrain, and hindbrain stained for laminin revealed glomeruloid bodies throughout the brain parenchyma. In the sections shown, the anterior forebrain section was at the beginning of the lateral ganglionic eminence. The posterior forebrain section was near its junction with the midbrain as indicated by the presence of the aqueduct of Sylvius, lateral ventricle, choroid plexus, and third ventricle. The hindbrain section was at the level of the aqueduct of Sylvius and fourth ventricle. (B) Magnification of the boxed areas in panel A. Anterior forebrain inset: glomeruloid bodies were present at the bottom of the brain entering from the pial vasculature and in the subventricular zone adjacent to the lateral ventricle (arrows) (scale bar, 60 µm). Posterior forebrain inset: glomeruloid bodies were present throughout the brain parenchyma and adjacent to areas of cell death (dotted black line). Large glomerular bodies originate from pial vessels adjacent to the lateral ventricle (arrow) (scale bar, 80 µm). Hindbrain inset: glomeruloid bodies were present throughout the parenchyma, and pial vessels at the pia-brain interface were disorganized (arrows) (scale bar, 100 µm). Glomeruloid bodies were present in the spinal cord (SC). Adjacent to the spinal cord is the vertebral body primordium (VBP) (scale bar, 100 µm). Corresponding coronal sections from a Tfpi+/+ littermate are presented in supplemental Figure 2.
Glomeruloid bodies were present in the forebrain, hindbrain, and spinal cord of E14 Tfpi−/−brain. (A) Coronal sections of anterior forebrain, posterior forebrain, and hindbrain stained for laminin revealed glomeruloid bodies throughout the brain parenchyma. In the sections shown, the anterior forebrain section was at the beginning of the lateral ganglionic eminence. The posterior forebrain section was near its junction with the midbrain as indicated by the presence of the aqueduct of Sylvius, lateral ventricle, choroid plexus, and third ventricle. The hindbrain section was at the level of the aqueduct of Sylvius and fourth ventricle. (B) Magnification of the boxed areas in panel A. Anterior forebrain inset: glomeruloid bodies were present at the bottom of the brain entering from the pial vasculature and in the subventricular zone adjacent to the lateral ventricle (arrows) (scale bar, 60 µm). Posterior forebrain inset: glomeruloid bodies were present throughout the brain parenchyma and adjacent to areas of cell death (dotted black line). Large glomerular bodies originate from pial vessels adjacent to the lateral ventricle (arrow) (scale bar, 80 µm). Hindbrain inset: glomeruloid bodies were present throughout the parenchyma, and pial vessels at the pia-brain interface were disorganized (arrows) (scale bar, 100 µm). Glomeruloid bodies were present in the spinal cord (SC). Adjacent to the spinal cord is the vertebral body primordium (VBP) (scale bar, 100 µm). Corresponding coronal sections from a Tfpi+/+ littermate are presented in supplemental Figure 2.
The pia mater vasculature was disorganized in E15.5 Tfpi−/− embryos
The specificity of the vascular defects for the CNS suggested a role of TFPI on the unique angiogenic development of vasculature in the brain and spinal cord.25 Therefore, vessel sprouting and growth from the perineural vascular plexus (PNVP) was examined. Immunohistochemical staining for laminin highlighted differences between the well-organized vasculature invading from the PNVP into the brain parenchyma in the Tfpi+/+ embryo and the irregular architecture of the invading vessels in the Tfpi−/− embryo. The Tfpi−/− vasculature developed glomeruloid bodies immediately adjacent to the pia mater as vessels penetrated the brain parenchyma (Figure 4A-B). Abnormal meningeal development also was evident within the Tfpi−/− embryos. All 3 layers of the meninges were present, but in some instances, the layers were less defined than those observed in Tfpi+/+ littermates, suggesting a possible delay in their growth and development (Figure 4B). In areas with abnormal pia mater vasculature, the vasculature of the arachnoid mater also appeared developmentally delayed with poor definition of its 2 layers (Figure 4A-B).
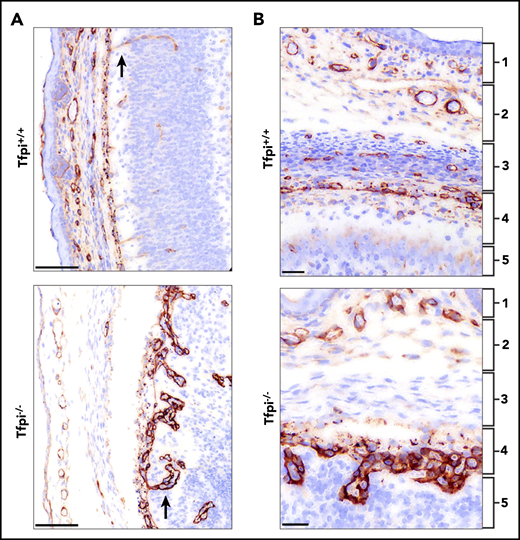
The pial vessels in the Tfpi−/−embryos were disorganized and produced glomeruloid bodies upon penetrating into the brain parenchyma. Meninges and pial vessels of E16 Tfpi+/+ and Tfpi−/− littermates stained for laminin. (A) The meninges of the Tfpi+/+ brain are well organized with normal vasculature seen entering the brain parenchyma (arrow). A prominent feature of the Tfpi−/− meninges is the large layer of abnormal vascular endothelial cells piled up in the pia mater that formed glomeruloid bodies upon entering the brain parenchyma (arrow) (scale bars, 100 µm). (B) The meningeal layers are identified by numbered brackets: (1) skeletogenous layer of the future skull, (2) dura mater, (3) arachnoid mater, (4) pia mater, and (5) brain parenchyma. Formation of the meninges was delayed in Tfpi−/− embryos when compared with Tfpi+/+ littermates. Vasculature was absent in the arachnoid layer of Tfpi−/− embryos, which lacks definition of the impermeable outer layer that prevents passage of cerebrospinal fluid from the subarachnoid space (scale bars, 30 µm).
The pial vessels in the Tfpi−/−embryos were disorganized and produced glomeruloid bodies upon penetrating into the brain parenchyma. Meninges and pial vessels of E16 Tfpi+/+ and Tfpi−/− littermates stained for laminin. (A) The meninges of the Tfpi+/+ brain are well organized with normal vasculature seen entering the brain parenchyma (arrow). A prominent feature of the Tfpi−/− meninges is the large layer of abnormal vascular endothelial cells piled up in the pia mater that formed glomeruloid bodies upon entering the brain parenchyma (arrow) (scale bars, 100 µm). (B) The meningeal layers are identified by numbered brackets: (1) skeletogenous layer of the future skull, (2) dura mater, (3) arachnoid mater, (4) pia mater, and (5) brain parenchyma. Formation of the meninges was delayed in Tfpi−/− embryos when compared with Tfpi+/+ littermates. Vasculature was absent in the arachnoid layer of Tfpi−/− embryos, which lacks definition of the impermeable outer layer that prevents passage of cerebrospinal fluid from the subarachnoid space (scale bars, 30 µm).
Glomeruloid bodies were present in E12.5 Tfpi−/− embryos
The embryonic brain undergoes active angiogenesis at E12.5 when vessels sprouting from the PNVP invade the neocortex and migrate radially toward the subventricular zone.26 The brains from Tfpi−/− embryos were examined at E12.5 for vascular pathology during this critical time of cortical angiogenesis (supplemental Figure 4). Hyperplasia of the pial vasculature with adjacent abnormal vasculature was present within the brain parenchyma (supplemental Figure 4B-C). Glomeruloid bodies were localized near the wall of the fourth ventricle and within the midbrain region, where they surrounded an area of cellular death (supplemental Figure 4B). Thus, the lack of TFPI is associated with defects in the early stages of cerebral vascular angiogenesis initiated from the PNVP.
Pericytes and microglial cells were localized to glomeruloid bodies in the Tfpi−/− embryo
The cerebral vasculature has more pericytes than other vascular beds,27,28 and pericytes stained with PDGFR-β were readily observed lining cerebral vessels in the Tfpi+/+ embryo (Figure 5A). Abundant pericytes were associated with endothelial cells of glomeruloid bodies in Tfpi−/− embryos (Figure 5A). Interestingly, the staining pattern for PDGFR-β was more localized and punctate in pericytes within glomeruloid bodies than in pericytes of normal vasculature. This altered staining pattern is perhaps another consequence of the abnormal vascular development within the glomeruloid body, although its significance remains to be determined. Microglial cells enter the brain between E8.5 and E9.5, are the only resident immune cell found in the healthy brain parenchyma, and persist in the brain throughout adulthood with self-renewal capabilities independent of hematopoietic progenitor cells.29,30 Microglia were examined in Tfpi−/− embryos because they play an important role in angiogenesis within the brain, including sprouting, migration, anastomosis, and vascular refinement.31 Microglial cells localized to both normal vasculature in the Tfpi+/+ embryos and the glomeruloid bodies in Tfpi−/− embryos, with no major differences in appearance or abundance (Figure 5B).
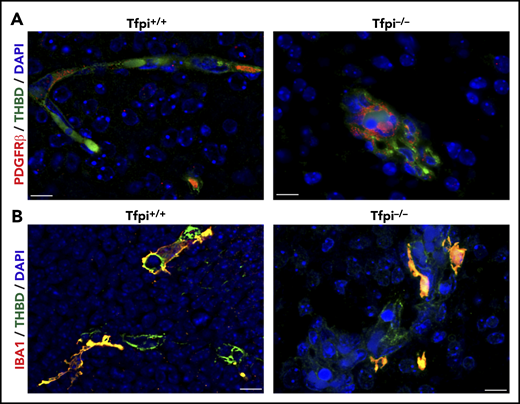
Pericytes and microglial cells were present within glomeruloid bodies in the E15.5 Tfpi−/−brain. (A) Immunofluorescence of brain vasculature from littermates stained for PDGFR-β (red) and thrombomodulin (green), with DAPI-stained nuclei (blue). Occasional pericytes (red) were present along vasculature within the Tfpi+/+ brain parenchyma. The glomeruloid bodies within the Tfpi−/− embryo had numerous pericytes associated with the aggregated endothelial cells (scale bars, 10 µm). (B) Immunofluorescence of brain vasculature from littermates stained for ionized calcium-binding adapter molecule 1 (IBA1; red/yellow) and thrombomodulin (green), with DAPI-stained nuclei (blue). IBA1-expressing microglial cells were associated with endothelial cells in Tfpi+/+ and Tfpi−/− embryos, with no discernible differences between genotypes (scale bars, 10 µm).
Pericytes and microglial cells were present within glomeruloid bodies in the E15.5 Tfpi−/−brain. (A) Immunofluorescence of brain vasculature from littermates stained for PDGFR-β (red) and thrombomodulin (green), with DAPI-stained nuclei (blue). Occasional pericytes (red) were present along vasculature within the Tfpi+/+ brain parenchyma. The glomeruloid bodies within the Tfpi−/− embryo had numerous pericytes associated with the aggregated endothelial cells (scale bars, 10 µm). (B) Immunofluorescence of brain vasculature from littermates stained for ionized calcium-binding adapter molecule 1 (IBA1; red/yellow) and thrombomodulin (green), with DAPI-stained nuclei (blue). IBA1-expressing microglial cells were associated with endothelial cells in Tfpi+/+ and Tfpi−/− embryos, with no discernible differences between genotypes (scale bars, 10 µm).
Glomeruloid bodies did not form in Tfpi−/− embryos lacking FV
In mice, the procoagulant cofactor, FV, is produced in distinct plasma and platelet pools.20 F5−/− embryos can survive to adulthood with transgenes that specifically express FV in either the liver, to produce plasma FV (F5LiverTg), or megakaryocytes, to produce platelet FV (F5PltTg).20 We have previously observed that Tfpi−/− embryos survive to adulthood when they completely lack FV expression at their endogenous locus (F5−/−) while expressing either plasma or platelet FV via F5LiverTg or F5PltTg.32 Therefore, we considered a potential role for the interaction between TFPI and FV in cerebrovascular development. F5−/−Tfpi−/− mice carrying F5LiverTg or F5PltTg were crossed with F5+/−Tfpi+/− mice for timed matings with embryonic harvest at ∼E15.5. Four litters from each type of transgenic mating were collected, containing a total of 53 embryos. A pathologist blinded to the genotype of the embryos examined the brains for the presence of glomeruloid bodies and associated pathology. In each of the 21 F5+/−Tfpi+/− embryos, brain tissue appeared normal. In contrast, each of the 11 F5+/−Tfpi−/− embryos, regardless of FV transgenic line, were histologically indistinguishable from Tfpi−/− embryos when evaluated with trichrome, fibrinogen, and laminin staining (Figures 6A-B), confirming the 100% penetrance of this phenotype in Tfpi−/− embryos. In contrast, glomeruloid bodies and associated brain pathology were totally absent in the 5 F5−/−Tfpi−/− embryos that did not express a FV transgene, as they had normal vasculature within the brain parenchyma and pia-brain interface (Figures 6A-B). This indicates that FV is required for the development of glomeruloid bodies in Tfpi−/− embryos.
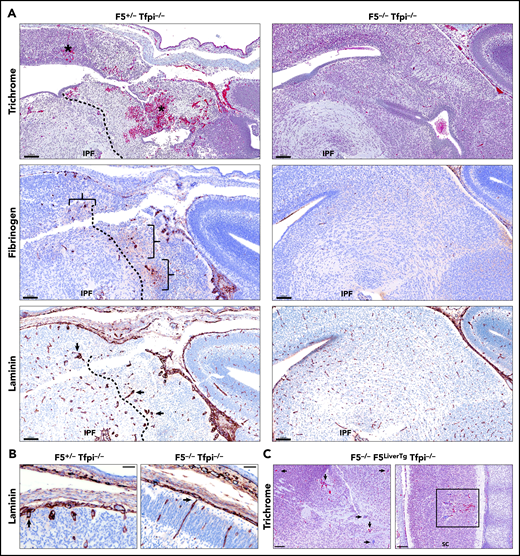
E15.5 Tfpi−/−embryos lacking FV had normal cerebral vasculature. (A) Trichrome-stained midbrain from a F5+/−Tfpi−/− embryo had a large area of hemorrhage (asterisk) and necrosis surrounded by glomeruloid bodies, essentially identical to that observed in Tfpi−/− embryos. In contrast, F5−/−Tfpi−/− embryos had normal brain vasculature without evidence of glomeruloid bodies, hemorrhage, or cellular death. Glomeruloid bodies were seen surrounded by fibrinogen (brackets) leaking from the abnormal vessels. The dotted line delineated the brain tissue with massive cell death (right) from that of the living cells (left). Normal architecture of the F5−/−Tfpi−/− embryo brain displayed normal vasculature without fibrinogen leakage. Laminin staining of the F5+/−Tfpi−/− brain revealed numerous glomeruloid bodies throughout the brain parenchyma (arrows point to 3 glomeruloid bodies). The dotted line delineates cell death (right) from living cells (left). The F5−/−Tfpi−/− brain had normal vasculature with laminin-stained basement membrane. Scale bars, 100 µm. (B) Trichrome stain of pial vessels (arrows) penetrating the brain parenchyma in the F5+/−Tfpi−/− embryo (left) and F5−/−Tfpi−/− embryo (right). The penetrating vasculature of the F5+/−Tfpi−/− embryo revealed glomeruloid bodies advancing from the disorganized pia mater, while the vasculature from the F5−/−Tfpi−/− embryo is organized with parallel walls (scale bar, 50 µm). (C) Trichrome-stained sections from F5−/−Tfpi−/− embryos carrying the F5LiverTg. Rare glomeruloid bodies were observed in the midbrain of one embryo (arrows) and spinal cord of another embryo (sc; square). Minor hemorrhage is present in the spinal cord but is not associated with cell death or inflammation. No pathology was observed in 3 other F5−/−F5LiverTgTfpi−/− embryos (scale bar, 100 µm).
E15.5 Tfpi−/−embryos lacking FV had normal cerebral vasculature. (A) Trichrome-stained midbrain from a F5+/−Tfpi−/− embryo had a large area of hemorrhage (asterisk) and necrosis surrounded by glomeruloid bodies, essentially identical to that observed in Tfpi−/− embryos. In contrast, F5−/−Tfpi−/− embryos had normal brain vasculature without evidence of glomeruloid bodies, hemorrhage, or cellular death. Glomeruloid bodies were seen surrounded by fibrinogen (brackets) leaking from the abnormal vessels. The dotted line delineated the brain tissue with massive cell death (right) from that of the living cells (left). Normal architecture of the F5−/−Tfpi−/− embryo brain displayed normal vasculature without fibrinogen leakage. Laminin staining of the F5+/−Tfpi−/− brain revealed numerous glomeruloid bodies throughout the brain parenchyma (arrows point to 3 glomeruloid bodies). The dotted line delineates cell death (right) from living cells (left). The F5−/−Tfpi−/− brain had normal vasculature with laminin-stained basement membrane. Scale bars, 100 µm. (B) Trichrome stain of pial vessels (arrows) penetrating the brain parenchyma in the F5+/−Tfpi−/− embryo (left) and F5−/−Tfpi−/− embryo (right). The penetrating vasculature of the F5+/−Tfpi−/− embryo revealed glomeruloid bodies advancing from the disorganized pia mater, while the vasculature from the F5−/−Tfpi−/− embryo is organized with parallel walls (scale bar, 50 µm). (C) Trichrome-stained sections from F5−/−Tfpi−/− embryos carrying the F5LiverTg. Rare glomeruloid bodies were observed in the midbrain of one embryo (arrows) and spinal cord of another embryo (sc; square). Minor hemorrhage is present in the spinal cord but is not associated with cell death or inflammation. No pathology was observed in 3 other F5−/−F5LiverTgTfpi−/− embryos (scale bar, 100 µm).
The absence of platelet FV prevented glomeruloid body formation in Tfpi−/− embryos
The F5PltTg produces ∼5% normal platelet FV and no plasma FV.20 No glomeruloid bodies were present in the brain of the single F5−/−F5PltTgTfpi−/− embryo present among the 53 obtained from the above crosses. The only lesion identified was a very small microhemorrhage. F5LiverTg produces ∼45% normal levels of plasma FV and no platelet FV.20 Five of the 53 embryos were F5−/−Tfpi−/− and carried the F5LiverTg. These embryos had either no or rare glomeruloid bodies in the brain (Figure 6C). Since all the F5+/−Tfpi−/− embryos had glomeruloid body formation with severe secondary brain pathology and F5−/−F5LiverTg mice primarily differ from F5+/− mice in that they do not produce platelet FV, this finding suggests that lack of platelet FV ameliorates the formation of glomeruloid bodies in the brain of Tfpi−/− embryos.
Gpr124 was expressed in vasculature of glomeruloid bodies
The absence of glomeruloid bodies and associated brain pathology in Tfpi−/− embryos lacking FV suggests that excess thrombin produced by prothrombinase drives the phenotype. Mice with global or endothelial-specific deletion of Gpr124 (also called tumor endothelial marker 5) succumb to embryonic lethality with abnormal angiogenesis and production of glomeruloid bodies in the forebrain and spinal cord that mimics the vascular phenotype observed in Tfpi−/− embryos.33 Thrombin has been reported to cleave an N-terminal 60-kDa fragment of the extracellular domain of Gpr124.34 Therefore, we hypothesized that excess thrombin production in Tfpi−/− embryos results in proteolytic inactivation of Gpr124, which contributes to the similar phenotypes observed in these 2 mouse models. Vessel sprouting and growth from the PNVP of Tfpi+/+ and Tfpi−/− littermates were examined using dual-fluorescent staining for the extracellular domain of Gpr124 and thrombomodulin (Figure 7). Abundant staining of Gpr124 was observed within all vessels, including those in glomeruloid bodies, suggesting that degradation of Gpr124 is not responsible for the production of glomeruloid bodies in Tfpi−/− mice.
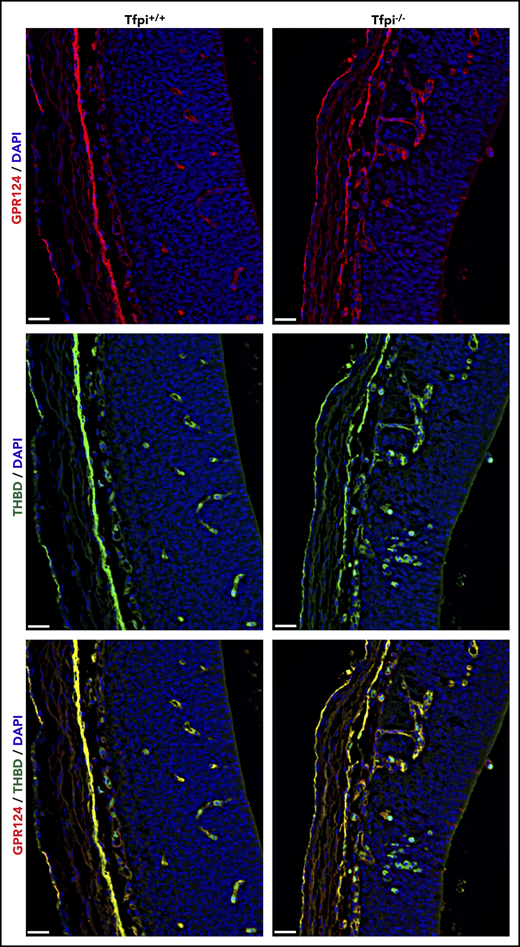
Gpr124 was present in glomeruloid bodies of E13.5 Tfpi−/−brain. Forebrain sections from Tfpi+/+ and Tfpi−/− littermates were stained for the extracellular domain of Gpr124 (red) and thrombomodulin (green), with DAPI-stained nuclei (blue) to depict the PNVP of the anterior forebrain and the adjacent lateral ventricle. Gpr124 stains well-organized vasculature in the Tfpi+/+ brain and the disorganized vasculature penetrating from the PNVP into the brain parenchyma in the Tfpi−/− brain. In the overlay, thrombomodulin stains the endothelial cells in the same pattern as Gpr124 in both embryos. Autofluorescence of the embryonic nucleated RBCs is observed within the vasculature of both embryos and areas of hemorrhage in the Tfpi−/− embryo (scale bars, 30 µm).
Gpr124 was present in glomeruloid bodies of E13.5 Tfpi−/−brain. Forebrain sections from Tfpi+/+ and Tfpi−/− littermates were stained for the extracellular domain of Gpr124 (red) and thrombomodulin (green), with DAPI-stained nuclei (blue) to depict the PNVP of the anterior forebrain and the adjacent lateral ventricle. Gpr124 stains well-organized vasculature in the Tfpi+/+ brain and the disorganized vasculature penetrating from the PNVP into the brain parenchyma in the Tfpi−/− brain. In the overlay, thrombomodulin stains the endothelial cells in the same pattern as Gpr124 in both embryos. Autofluorescence of the embryonic nucleated RBCs is observed within the vasculature of both embryos and areas of hemorrhage in the Tfpi−/− embryo (scale bars, 30 µm).
Discussion
We have demonstrated that Tfpi−/− mouse embryos have stunted brain growth and delayed meningeal development during late gestation. The stunted brain growth was associated with severe vascular pathology in the parenchyma of the forebrain, midbrain, hindbrain, and spinal cord characterized by glomeruloid bodies surrounding areas of cellular death, fibrin deposition, and hemorrhage. Thus, absence of the K1 domain of the anticoagulant protein TFPI is associated with a tissue-specific cerebrovascular phenotype that may contribute to the late gestational embryonic death of Tfpi−/− embryos.
Tfpi−/− embryos also had abnormally developed meninges during late gestation with incomplete demarcation of the vascular and connective tissue layers of the arachnoid mater, disorganized vasculature within the pia mater, and formation of glomeruloid bodies upon vascular penetration from the PNVP into the adjacent brain parenchyma. Unlike other tissues that create vessels via vasculogenesis, the brain develops its vascular bed by angiogenic sprouting from the PNVP followed by extensive pruning and regression to form the final functional vasculature.25,35,36 Since this activity of TFPI was specific to the CNS, and there was normal vascular development within other tissues, our findings suggest that TFPI modulates vascular development occurring through angiogenesis, but not through vasculogenesis.
Pericytes and microglial cells migrate from the yolk sac to the CNS before neurovascular development. They play an important role in angiogenesis and blood-brain barrier integrity and contribute to the production and deposition of laminin into the basement membrane.37-39 Thus, they provide vascular support during development and express TFPI.40,41 The Tfpi−/− embryonic brain had both cell types present, indicating that the vascular pathology was not due to the absence of either of these critical cell lineages.
FV is a cofactor for FXa in prothrombinase, the proteolytic complex responsible for converting prothrombin to thrombin in the coagulation cascade. Development of pathology within the brain of late-gestation Tfpi−/− embryos required the presence of FV, as the vascular lesions and associated pathology were completely absent in Tfpi−/− embryos lacking endogenous FV. This striking finding strongly suggests that TFPI dampens FV-dependent production of thrombin in a manner that modulates cerebrovascular development. Interestingly, platelet FV appeared to influence the phenotype, as F5+/−Tfpi−/− embryos, which expressed 50% FV levels in both the plasma and the platelet, developed severe brain pathology, while F5−/−Tfpi−/− embryos carrying F5LiverTg, which have 45% plasma FV and no platelet FV, had normal brain development and cerebral vasculature, with rare vascular defects observed in 2 of 5 embryos. Therefore, the effect of TFPI on cerebral vasculature development appears to be mediated through its ability to diminish the generation of excess thrombin activity rather than directly inhibit the upstream coagulation catalytic complexes, TF-FVIIa or TF-FVIIa-FXa, which mediate signaling events through PAR1 and PAR2 that modulate tumor angiogenesis.8,9,42,43
Although the glomeruloid body CNS phenotype requires the presence of FV, the CNS has higher amounts of TF than other tissues.44 Thus, TFPI may diminish thrombin generation in the developing brain through its inhibition of TF-FVIIa activity and consequent dampening of downstream thrombin production,1 a concept supported by the normal appearance of E17.5 Tfpi−/− embryos lacking FVII.11 Alternatively, TFPI also directly inhibits early prothrombinase activity, which is mediated, in part, by a tight-binding interaction between a basic region near the C terminus of TFPIα and an acidic region in the B-domain of FV.12,14 Support of a role for direct inhibition of prothrombinase by TFPI in modulation of vascular development is provided by previous findings showing the C-terminal region of TFPIα inhibits proliferation of cultured endothelial cells45,46 and angiogenesis in a murine hindlimb model.47 Further, there are multiple, well-characterized differences in the production and tissue expression of TFPI48,49 and FV20,50 between mice and humans. Additional studies are needed to specifically define how different TFPI anticoagulant activities modulate thrombin production in the developing brain and how our findings in mice may apply to cerebrovascular development in humans.
Thrombin is a major activator of PAR receptors, which are expressed in developing vasculature.51,52 Indeed, it has been suggested that developing vasculature senses thrombin and other coagulation factors in a manner that orchestrates the formation and remodeling of embryonic blood vessels.6 Thus, altered activation of endothelial PARs by excess thrombin may contribute to the vascular development defects observed in Tfpi−/− embryos in a manner that is not well understood. However, the phenotype observed in the Tfpi−/− embryos is similar in several aspects to that observed in embryos lacking components of β-catenin signaling pathways, including Gpr124, where development of glomeruloid bodies and associated pathology have been extensively documented in the embryonic brain with normal vascular development in other tissues.25,33,53 Therefore, we considered the possibility that the absence of TFPI and consequent excess thrombin production alters β-catenin signaling within the developing brain.
The 2 principal effectors of β-catenin signaling that control cerebral vascular development are Wnt7a/b or Norrin binding to frizzled/low-density lipoprotein receptor-related protein (LRP) receptor complexes.54 Wnt7a/7b interacts with Gpr124, Lrp5/6, and Reck to promote vascular development predominantly in the forebrain and spinal cord, while Norrin interacts with tetraspanin 12 and Lrp5 to promote developmental angiogenesis predominantly in the hindbrain and spinal cord.54 Deficiency of components of these pathways in mice produce CNS-specific vascular defects that closely mimic some features observed in Tfpi−/− embryos but differ in the spatial location of glomeruloid body formation. For example, the vasculature in Tfpi−/− embryos was affected in the forebrain, midbrain, hindbrain, and spinal cord compared with defects in the β-catenin pathway featuring localized effects in the embryonic forebrain (Wnt7a/7b) or hindbrain (Norrin). This suggested the possibility that excess thrombin produced in Tfpi−/− embryos may degrade one or more of the extracellular effectors of the β-catenin signaling complexes. In support of this hypothesis, it has been reported that thrombin degrades Gpr124 by cleaving at 2 specific sites within its extracellular domain.34 However, staining for the extracellular domain of Gpr124 in the brain of Tfpi−/− embryos revealed the presence of Gpr124 on normal-appearing vasculature and vessels within glomeruloid bodies. Further studies investigating how coagulation protease activity may intersect with effectors of β-catenin signaling pathways will enhance our understanding of the complex pathways regulating cerebral vascular development and define potential links responsible for the similar defects in cerebrovascular development observed in the embryos lacking TFPI and components of the β-catenin pathway.
In summary, TFPI is an inhibitor of TF-FVIIa and prothrombinase, 2 of the main catalytic complexes of the procoagulant response.49 The presence of severe CNS vascular anomalies in Tfpi−/− embryos implicates the coagulation system as a key player in angiogenic signaling pathways within the developing CNS in mice. Since the CNS pathology was fully rescued by the absence of FV, the modulation of thrombin production by TFPI likely drives the phenotype.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The mouse lines used in this study are available publically. The authors will provide other reagents and protocols to other investigators upon request.
Acknowledgments
The authors wish to thank Barbara Fleming for expert assistance in preparing histology slides.
This work was supported by National Institutes of Health (NIH) grants HL068835 (A.E.M.), HL135035 (R.J.W.), HL135793 (D.G.), HL117132 (H.W.), HL133348 (H.W.), and HL130678 (H.W.) from the National Heart, Lung, and Blood Institute (NHLBI), and AI133561 (H.W.) from the National Institute on Aging; a research grant from Novo Nordisk (A.E.M.); and American Heart Association innovative research grant 17IRG33460238 (R.J.W.). D.G. is an investigator of the Howard Hughes Medical Institute. A.E.S. was supported by NIH training grant HL007209 from the NHLBI.
Authorship
Contribution: S.A.M. and R.J.W. designed research, performed research, analyzed data, and wrote the paper; A.C.C. performed research and wrote the paper; N.D.M. and M.Z. performed research; A.E.S., D.G., and H.W. analyzed data and wrote the paper; and A.E.M. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: A.E.M. receives research funding from Novo Nordisk and has received honoraria for serving on Novo Nordisk Advisory Boards. The remaining authors declare no competing financial interests.
Correspondence: Alan E. Mast, Versiti Blood Research Institute, PO Box 2178, Milwaukee, WI 53201-2178; e-mail: aemast@versiti.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal