

Key Points
Fibrin(ogen) deposition in the injured liver does not depend on traditional hemostatic mechanisms.
FXIII-driven fibrin(ogen) cross-linking does not require fibrin polymerization in the acutely injured liver.
Visual Abstract

Abstract
Intravascular fibrin clot formation follows a well-ordered series of reactions catalyzed by thrombin cleavage of fibrinogen leading to fibrin polymerization and cross-linking by factor XIIIa (FXIIIa). Extravascular fibrin(ogen) deposits are observed in injured tissues; however, the mechanisms regulating fibrin(ogen) polymerization and cross-linking in this setting are unclear. The objective of this study was to determine the mechanisms of fibrin polymerization and cross-linking in acute liver injury induced by acetaminophen (APAP) overdose. Hepatic fibrin(ogen) deposition and cross-linking were measured following APAP overdose in wild-type mice, mice lacking the catalytic subunit of FXIII (FXIII−/−), and in FibAEK mice, which express mutant fibrinogen insensitive to thrombin-mediated fibrin polymer formation. Hepatic fibrin(ogen) deposition was similar in APAP-challenged wild-type and FXIII−/− mice, yet cross-linking of hepatic fibrin(ogen) was dramatically reduced (>90%) by FXIII deficiency. Surprisingly, hepatic fibrin(ogen) deposition and cross-linking were only modestly reduced in APAP-challenged FibAEK mice, suggesting that in the APAP-injured liver fibrin polymerization is not strictly required for the extravascular deposition of cross-linked fibrin(ogen). We hypothesized that the oxidative environment in the injured liver, containing high levels of reactive mediators (eg, peroxynitrite), modifies fibrin(ogen) such that fibrin polymerization is impaired without impacting FXIII-mediated cross-linking. Notably, fibrin(ogen) modified with 3-nitrotyrosine adducts was identified in the APAP-injured liver. In biochemical assays, peroxynitrite inhibited thrombin-mediated fibrin polymerization in a concentration-dependent manner without affecting fibrin(ogen) cross-linking over time. These studies depict a unique pathology wherein thrombin-catalyzed fibrin polymerization is circumvented to allow tissue deposition and FXIII-dependent fibrin(ogen) cross-linking.
Introduction
Intravascular formation of cross-linked fibrin polymers (ie, fibrin clots) occurs through a series of reactions driven by the coagulation protease thrombin. Fibrinogen is a hexameric plasma protein comprised of 2 Aα, Bβ, and γ chains. Thrombin cleaves fibrinopeptides on the Aα and Bβ chain of the fibrinogen molecule. This enables α-γ and β-β chain knob-hole interactions, promoting the spontaneous formation of fibrin polymers.1 These initial steps drive proteolytic consumption of plasma fibrinogen and deposition of fibrin polymer at sites of vascular injury or within pathologic intravascular thrombi. The transglutaminase coagulation factor XIII (FXIII) circulates in complex with fibrinogen, and on activation, cross-links the α and γ chains of fibrin molecules, generating γ-γ dimers and other higher-molecular-weight products (eg, α polymers). Importantly, these covalent bonds formed by FXIIIa play a critical role in defining the structure, stability, and effector functions of fibrin in both hemostasis and thrombosis.2
Thrombin-catalyzed fibrin polymer formation precedes cross-linking in traditional coagulation reactions essential for hemostasis and thrombosis.1 Indeed, thrombin-mediated fibrin polymer formation is widely perceived as a prerequisite for FXIIIa-driven cross-linking in vivo. Indeed, fibrin polymerization by thrombin promotes activation of FXIIIa during clot formation, ensuring that the developing clot has an appropriate supply of FXIII during its formation.3 Interestingly, prior studies have shown that under certain conditions in vitro (ie, supraphysiologic Ca2+ concentrations, presence of reducing thiol chemicals), even without thrombin cleavage and fibrinopeptide release, FXIIIa can form cross-linked complexes of soluble fibrinogen,4-6 although at approximately an eightfold slower rate than polymerized fibrin.6 It remains unclear, however, whether FXIIIa can cross-link soluble fibrinogen in the absence of fibrin polymerization in vivo, and if so, what physiologic or pathophysiologic mechanisms could drive this alternative pathway.
One reason for the knowledge gap regarding in vivo FXIIIa cross-linked fibrinogen may be because of the focus within the field on mechanisms driving fibrin polymerization and cross-linking in the context of intravascular coagulation. However, extravascular deposition of cross-linked fibrin(ogen) is evident in pathologic conditions, such as that observed within areas of tissue necrosis. For example, acute hepatic necrosis induced by overdose of the widely used over-the-counter drug acetaminophen (APAP) is associated with reductions in plasma fibrinogen and robust deposition of fibrin(ogen) throughout the necrotic zones within the injured liver.7-10 In experimental settings of acetaminophen-induced liver damage, fibrin(ogen) drives hepatoprotective processes in the APAP-injured liver,8 yet we know very little about the mechanisms controlling fibrin(ogen) deposition and cross-linking in the injured liver.
In this study, through rigorous analysis of intrahepatic fibrin(ogen) deposits and combined application of FXIII-deficient mice and novel mice (FibAEK mice) expressing a mutant form of fibrinogen insensitive to thrombin cleavage (fibrinogenAEK),11 we sought to define the mechanisms of fibrin(ogen) polymerization and cross-linking in the acutely injured liver.
Materials and methods
Mice
Mice expressing a mutant form of fibrinogen Aα chain insensitive to thrombin cleavage (FibAEK mice) and mice lacking the factor XIII catalytic A subunit (FXIII−/− mice) have been described previously.11,12 Male mutant mice and wild-type (WT) mice matched for C57Bl/6J background were used in age-matched cohorts between the ages of 10 and 20 weeks. Data interpretation for in vivo studies were based on 2 matched cohorts of APAP-challenged WT vs FXIII−/− mice, 3 matched cohorts of APAP-challenged WT vs FibAEK mice, and matched cohorts of mice of each genotype treated with saline, performed at independent times, with end point analyses performed collectively. Mice were housed in Association for Assessment and Accreditation of Laboratory Animal Care–approved facilities at Michigan State University (MSU) at an ambient temperature of 22 ± 2°C with alternating 12-hour light/dark cycles and provided ad libitum access to standard rodent diet (Teklad 8940; Envigo, Indianapolis, IN) and purified drinking water. All animal procedures were approved by the MSU Institutional Animal Care and Use Committee.
Acute liver injury induced by APAP challenge, determination of liver injury, and labeling of hepatic fibrin(ogen)
Mice were challenged with APAP (300 mg/kg, intraperitoneally) or vehicle (sterile saline) at 30 μL/g body weight as described previously.7-10 Liver injury and hepatocellular necrosis were determined using plasma alanine aminotransferase activity and histologic analysis of hematoxylin and eosin (H&E)-stained paraffin-embedded liver sections as described previously.8,9 Hepatic fibrin(ogen) deposition was detected by immunolabeling of formalin-fixed paraffin-embedded or frozen liver sections as described previously.8,9 For full details of these methods, see supplemental Methods, available on the Blood Web site.
Detection of intrahepatic fibrin(ogen) by capillary western blotting
Hepatic levels of cross-linked fibrin(ogen) were measured as described previously.13 For full details, see supplemental Methods.
Peroxynitrite modification of fibrinogen
Human fibrinogen was modified with peroxynitrite (PN) as described previously with minor modifications.14 Briefly, purified human fibrinogen (Fib 1; Enzyme Research Laboratories, South Bend, IN) was diluted to 5.88 µM in 0.1 M Tris-HCl (pH 7.4), and 1 mL fibrinogen solution was added to 5-mL microcentrifuge tubes. Next, PN (MilliporeSigma, Burlington, MA) was diluted in 0.1 M NaOH and added to the tubes containing fibrinogen solution (0-1000 µM final concentration of PN) by pipetting a 2.75-µL drop on the side of the tube (slightly above the meniscus of the fibrinogen solution) and immediately vortexing for 15 seconds. The workflow for vehicle (0.1 M NaOH)-treated fibrin(ogen) was identical, and vehicle-treated fibrinogen is referred to as 0 µM PN henceforth. Experiments were performed as 3 independent replicates. Clot turbidity analysis and results from modified Clauss assay represent 1 of these replicates.
Assessment of thrombin-mediated fibrin polymerization
PN- or vehicle-modified fibrinogen (prepared as described above) was diluted to 1 mg/mL fibrinogen in 0.1 M Tris HCl (pH 7.4), and a 160-µL sample was added to each well of a 96-well microtiter plate (Greiner Bio-One, Monroe, NC). A solution of human thrombin (Enzyme Research Laboratories) and CaCl2 in 0.1 M Tris HC1 was rapidly added using a multichannel pipette to a final concentration of 0.25 U/mL thrombin and 5 mM CaCl2. Fibrin polymerization, monitored as a change in turbidity (absorbance at 595 nm), was measured every 25 seconds for 30 minutes using an Infinite M200 (Tecan, Mӓnnedorf, Switzerland) plate reader. Each assay was performed in triplicate.
Separately, the time required for PN- or vehicle-modified fibrinogen to clot was determined using a STart4 Coagulation Analyzer (Diagnostica Stago, Parsippany, NJ) and a modified Clauss fibrinogen assay. Briefly, 200 µL of fibrinogen sample diluted to 1 mg/mL in 0.1 M Tris-HCl was added to each cuvette and prewarmed to 37°C. Human thrombin (0.25 U/mL + 5 mM CaCl2 final concentration; Enzyme Research Laboratories) was then added, and the time to clot formation was recorded. Each fibrinogen sample was assayed in duplicate alongside an identical unmodified purified human fibrinogen control sample also in duplicate. The time to clot formation (measured in seconds) is presented as fold change compared with this control sample.
Determination of fibrin cross-linking by gel electrophoresis
PN- or vehicle-modified fibrinogen (prepared as described above) was diluted to 1 mg/mL in 0.1 M Tris-HCl. Thrombin (0.25 U/mL + 5 mM CaCl2 final concentration) was added and incubated for the indicated times at 37°C. The reaction was terminated by adding an equivalent volume of 2× Laemmli Sample Buffer (Bio-Rad Laboratories, Hercules, CA) containing 5% 2-mercaptoethanol (MilliporeSigma), vigorously vortexing, and heat denaturing at 95°C for 10 minutes. Samples were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 4% to 12% Bis-Tris gel (Bio-Rad) in running buffer containing 3-morpholinopropane-1-sulfonic acid (XT-MOPS; Bio-Rad). The gel was rinsed briefly in deionized water and stained for 30 minutes in SimplyBlue SafeStain total protein stain (Thermo Fisher Scientific, Waltham, MA), then destained through several changes of deionized water. The gel was digitized using an Odyssey CLx gel imaging system (LI-COR Biosciences, Lincoln, NE). Quantitation of fibrin(ogen) cross-linking was performed using Image Studio Lite Version 5.0 (LI-COR Biosciences).
Immunoprecipitation of nitrotyrosine-adducted proteins from liver
Male wild-type (C57Bl/6J) mice were challenged with 300 mg/kg APAP as described previously,8,9 and livers were collected 6 or 24 hours after APAP challenge. Immunoprecipitation was performed using a Pierce Classic Magnetic IP/CoIP Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Briefly, ∼30 mg of liver tissue was homogenized in Pierce IP Lysis Buffer containing 2× protease inhibitor cocktail (G BioSciences; St Louis, MO). Homogenates were diluted 1:4 in Pierce IP Lysis Buffer and incubated overnight with 10 µg/mL monoclonal anti-mouse 3-nitrotyrosine (3-NT) antibody (clone 39B6, #GTX41979; Genetex, Irvine, CA) at 4°C. Samples were then incubated with prewashed magnetic Protein A/G beads. Proteins were magnetically immunoprecipitated and eluted in the provided elution buffer followed by neutralization buffer.
Immunoblotting
For complete details of detection of nitrotyrosine residues in PN-modified purified human fibrinogen, fibrinogen in samples immunoprecipitated with anti-nitrotyrosine antibody, and fibrinogen in IP input samples, see supplemental Methods. Briefly, samples were diluted in Laemmli sample buffer containing 2-mercaptoethanol and heat denatured at 95°C for 10 minutes. Equal amounts of protein were loaded in a 4% to 12% Bis-Tris gel (Bio-Rad) and proteins were separated by separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis in XT-MOPS running buffer (Bio-Rad). Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (MilliporeSigma), using a Bio-Rad Transblot Semi-dry Transfer Cell, and the membrane was blocked 1 hour at room temperature. Following blocking, the membrane was incubated in blocking buffer containing primary antibody (anti-mouse nitrotyrosine, clone 39B6, Genetex or anti-human fibrinogen; Agilent Technologies, Santa Clara, CA) overnight at 4°C. Membranes were washed and incubated with the appropriate secondary antibody for 1 hour at room temperature.
For chemiluminescent detection, all membranes were incubated with Syngene SynPICO horseradish peroxidase substrate (Innovative Solutions, Beverly Hills, MI) and exposed to blue autoradiography film.
Scanning electron microscopy
Scanning electron microscopy (SEM) was performed by the MSU Center for Advanced Microscopy. Human fibrinogen was pretreated with PN or vehicle as described above. Clots were prepared by adding 0.5 mg/mL fibrinogen solution and human thrombin (Enzyme Research Laboratories) at a final concentration of 1 U/mL in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffered saline to the interior of a 1.75-mL Eppendorf tube cap. Before addition of sample, a needle was used to add a small hole to the cap to facilitate fluid exchange, as described previously.15 Samples were incubated for 60 minutes at 37°C in a humidified chamber. Samples were washed in 50 mM sodium cacodylate buffer, fixed in 3% glutaraldehyde, and dehydrated in ethanol solutions from 30% to 100% as described previously.11 Samples were critical point dried using carbon dioxide as a transitional fluid in a Leica Microsystems EM CPD300 (Leica Microsystems, Vienna, Austria). Samples were mounted on aluminum stubs using epoxy glue (System Three Resins, Inc, Auburn, WA), iridium coated, and examined using a JOEL 7500F scanning electron microscope (JOEL Ltd, Tokyo, Japan).
Statistical analyses
Results from mice from each independent study were analyzed collectively. Comparison of 2 groups was performed using a Student t test. Comparison of 4 groups was performed using 2-way analysis of variance and Student-Newman-Keuls post hoc test. In cases where data were not normally distributed, data were log or box-cox transformed to achieve a normal distribution before testing. Results were considered significant when P < .05. Analysis was performed using Prism (version 8; GraphPad Software, La Jolla, CA) and SigmaPlot (version 12.0; Systat Software, San Jose, CA).
Results
Fibrin(ogen) is cross-linked in the APAP-injured liver
APAP overdose caused acute hepatotoxicity in WT mice as indicated by centrilobular hepatic necrosis 24 hours after APAP challenge (Figure 1A, black arrowheads). In agreement with prior studies,8,9 hepatic fibrin(ogen) deposition was evident within areas of centrilobular hepatic necrosis (Figure 1B), whereas hepatic fibrin(ogen) deposition is routinely minimal in saline-treated mice (data not shown). Capillary-based western blotting using a polyclonal antibody against human fibrinogen identified high-molecular-weight (HMW) cross-linked fibrin(ogen) in insoluble liver extracts 24 hours after APAP challenge (Figure 1C). Using antibodies that uniquely detect the Aα, Bβ, and γ chains of mouse fibrinogen under reducing conditions (supplemental Figure 1), we observed that hepatic levels of Fibβ (∼51 kDa) increased dramatically in the liver after APAP challenge (Figure 1D,G). This finding was consistent with increased hepatic fibrin(ogen) labeling (Figure 1B). Using the Fibγ chain antibody, we detected an increase in peak area at a molecular weight (∼136 kDa) potentially reflecting γ-γ dimer after APAP challenge (Figure 1E,H). Because this peak appeared at a larger molecular weight than predicted for γ-γ dimer (∼93 kDa) using automated capillary western blotting, we attempted to further confirm the identity of this band using an anti–D-dimer antibody that reacts selectively with the fibrin(ogen) γ-γ cross-link catalyzed by FXIIIa (Zedira DD-XLink-mab A076). As anticipated, the anti–D-dimer antibody detected cross-linked Fibγ (eg, γ-γ dimer at ∼100 kDa) and fibrin degradation products somewhat selectively, as well as Fibγ (∼46 kDa) in purified human fibrinogen incubated with thrombin and/or plasmin (supplemental Figure 2A). These same fibrin(ogen) products were also detected at the same molecular weights using the anti-Fibγ antibody, which reacted with both purified mouse and human fibrin(ogen) (supplemental Figure 2B). However, no cross-reactivity with mouse fibrin(ogen) was observed using the anti–D-dimer antibody, which prevented us from using this antibody to detect fibrin(ogen) in mouse liver extracts. Based on these results, we will refer to this ∼136-kDa peak detected in liver extracts with automated capillary western blotting as cross-linked Fibγ. Higher-molecular-weight Fibγ peaks were also observed at approximately 200 and 380 kDa (Figure 1E). Importantly, we observed a peak at ∼46 kDa detected by the Fibγ antibody in a no-protein control sample (data not shown), suggesting cross-reactivity of the antibody with the fluorescent internal assay control included in each capillary; thus, peaks at this lower molecular weight do not reflect true Fibγ. Finally, the Fibα antibody detected a HMW peak (>300 kDa) in APAP-challenged mice, recapitulating observations with the polyclonal anti-fibrinogen antibody (Figure 1F,I).8 Collectively, the results indicate that cross-linking of the fibrinogen α and γ chains is evident in livers of APAP-challenged mice.
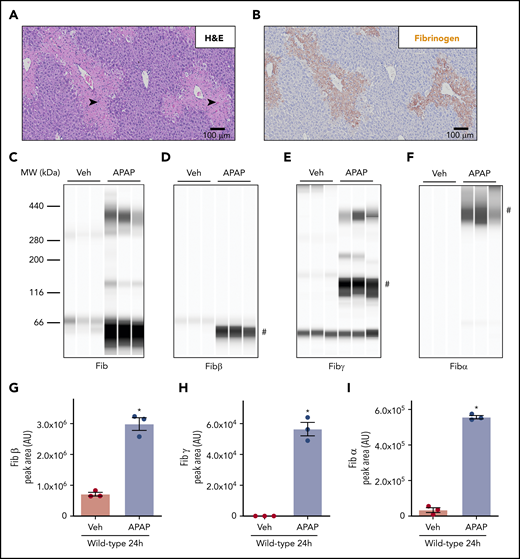
Deposition of cross-linked fibrin(ogen) in livers of mice after APAP overdose. WT mice were treated intraperitoneally with saline vehicle (Veh) or 300 mg/kg APAP, and hepatic fibrin(ogen) levels were determined 24 hours after challenge. (A-B) Representative photomicrographs (200×) show H&E-stained (A) and fibrin(ogen)-labeled (B, brown) liver sections. Necrosis denoted by black arrowhead. (C-I) Fibrin(ogen) levels were measured in enriched insoluble liver extracts using capillary-based western blotting (Wes). (C-F) Digital capillary images show fibrin(ogen) detected by rabbit polyclonal antibodies against fibrinogen (C) and selective for fibrin(ogen) Bβ (D), γ (E), and Aα chains (F). (G-I) Quantification of peaks indicated by the # in panels D-F is shown (n = 3 mice/group). Data are expressed as mean ± standard error of the mean. *Significantly (P < .05) different from saline-treated mice.
FXIII drives intrahepatic fibrin(ogen) cross-linking in the APAP-injured liver
In an intravascular clot, FXIIIa cross-links and stabilizes the polymerized fibrin network. Having determined that fibrin(ogen) deposits are cross-linked in the APAP-injured liver, we sought to determine the role of FXIII in hepatic fibrin(ogen) cross-linking. WT and FXIII−/− mice were challenged with APAP or vehicle. The average area of fibrin(ogen) labeling per field was quantitatively similar in WT and FXIII−/− mice 24 hours after APAP challenge (Figure 2A-B). To determine the role of FXIII in cross-linking of hepatic fibrin(ogen) deposits, we measured fibrin(ogen) levels in enriched insoluble protein fractions under reducing conditions. As anticipated based on fibrin(ogen) labeling in tissue sections (Figure 2A-B), levels of Fibβ increased equivalently in insoluble protein fractions from the livers of APAP-challenged WT and FXIII−/− mice (Figure 2C,F). Hepatic levels of cross-linked Fibγ and Fibα were increased in insoluble protein fractions from the livers of APAP-challenged WT mice (Figure 2D-E, G-H). Notably, levels of cross-linked Fibγ (∼136 kDa) were reduced to near-undetectable levels (Figure 2D,G) in APAP-challenged FXIII−/− mice. These findings were reproduced using standard immunoblotting for cross-linked Fibγ (∼120 kDa) in liver homogenates (supplemental Figure 3A). Moreover, cross-linked Fibα was reduced by approximately 90% in livers of APAP-challenged FXIII−/− mice compared with APAP-challenged WT mice (Figure 2E,H). Collectively, the results indicate that FXIII plays a major role in fibrin(ogen) cross-linking, but not fibrin(ogen) deposition, in the APAP-injured liver.
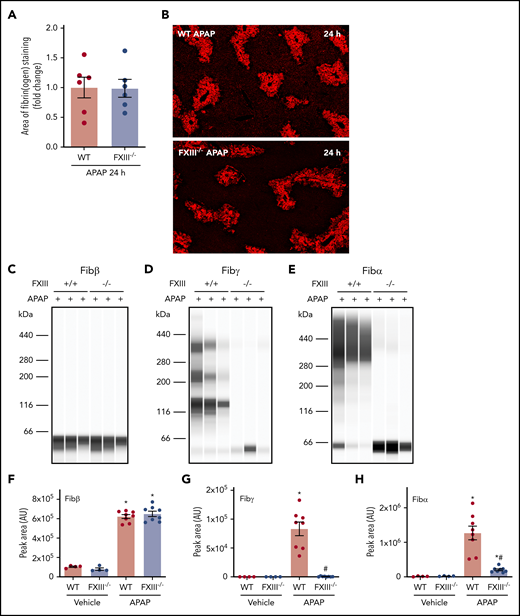
Impact of FXIII deficiency on hepatic fibrin(ogen) cross-linking after APAP overdose. FXIII−/− mice and WT mice were treated intraperitoneally with saline vehicle or 300 mg/kg APAP. Livers were collected 24 hours after challenge. (A) Fibrin(ogen) labeling in frozen liver sections was quantified as described in previously (n = 6 mice/group). (B) Representative photomicrographs of fibrinogen-labeled frozen sections (×5 virtual magnification). Fibrin(ogen) levels were measured in enriched insoluble liver extracts using capillary-based western blotting (Wes). (C-E) Representative digital capillary images show fibrin(ogen) detected and quantified (D-F) by rabbit polyclonal antibodies selective for fibrin(ogen) Aα, Bβ, and γ chains. n = 4 mice for vehicle-treated groups and 8 mice for APAP-challenged groups (F-H). Data expressed as mean ± standard error of the mean. *Significantly (P < .05) different from saline-treated mice of the same genotype. #Significantly (P < .05) different from APAP-challenged WT mice.
Prior studies have documented early hepatoprotective effects of fibrin(ogen) in mice challenged with a hepatotoxic dose of APAP.8 For instance, mice expressing fibrinogen that cannot engage β2 integrins develop increased necrosis and hepatic congestion/hemorrhage after APAP challenge.8 Thus, we determined whether the absence of FXIII-dependent fibrin(ogen) cross-linking affected APAP-induced liver injury. Interestingly, APAP hepatotoxicity was equivalent in WT and FXIII−/− mice, indicated by similar increases in plasma alanine aminotransferase (ALT) activity and hepatocellular necrosis 24 hours after APAP challenge (Figure 3A-C). The results indicate that FXIII-dependent fibrin(ogen) cross-linking is not central to the hepatoprotective effects of fibrin(ogen) 24 hours after APAP overdose.
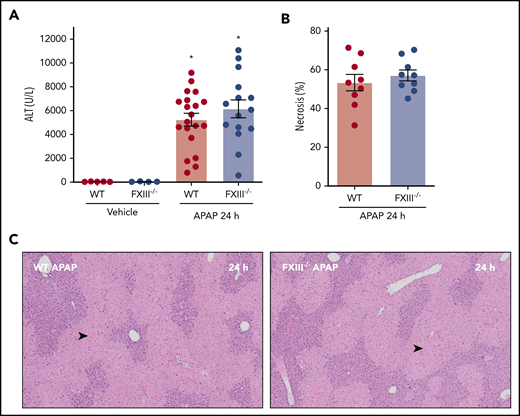
Impact of FXIII deficiency on liver injury after APAP overdose. FXIII−/− mice and WT mice were treated intraperitoneally with saline vehicle or 300 mg/kg APAP, and liver and citrated plasma were collected 24 hours after APAP challenge. (A) Plasma ALT activity was determined using commercial reagents (n = 4-5 mice/group for vehicle treated and 15-20 mice/group for APAP challenged). (B) Area of centrilobular necrosis was quantified as described (n = 9 mice/group). (C) Representative photomicrographs of H&E-stained liver sections (necrosis denoted by black arrowhead). Data expressed as mean ± standard error of the mean. *Significantly (P < .05) different from vehicle-treated mice of the same genotype.
Fibrin(ogen) cross-linking in the APAP-injured liver occurs in the absence of thrombin-catalyzed fibrin polymerization
Fibrin(ogen) accumulation in the APAP-injured liver is presumed to occur as a consequence of thrombin activity. Indeed, prior studies show that anticoagulants (eg, heparin) reduce hepatic fibrin(ogen) deposition early after APAP challenge.7 However, these drugs also reduce early hepatotoxicity.7,16 To selectively block thrombin-mediated fibrin polymerization in APAP-challenged mice, we used FibAEK mice, which express fibrinogen with mutant Aα chain fibrinopeptides fully resistant to cleavage by thrombin. As a result, fibrinogenAEK does not form fibrin polymers in response to thrombin.11
Fibrin(ogen) deposition increased in liver 24 hours after APAP challenge in WT mice compared with saline-challenged mice (Figure 4A-B). Hepatic fibrin(ogen) deposition tended to be lower in APAP-challenged FibAEK mice, indicated by immunofluorescent fibrin(ogen) labeling and measurement of Fibβ in insoluble extracts (Figure 4A-C). However, remarkable variation was observed, and neither measure achieved statistical significance. Because thrombin-catalyzed fibrin polymer formation is a prerequisite for cross-linking by FXIIIa under physiologic conditions,4,5 we anticipated a dramatic reduction in fibrinogen cross-linking in livers of APAP-challenged FibAEK mice comparable to FXIII−/− mice (Figure 2). To our surprise, hepatic levels of cross-linked fibrin(ogen), detected by both Fibα and Fibγ antibodies, were only modestly reduced in insoluble protein extracts from livers of APAP-challenged FibAEK mice, with the perceived reduction in Fibα not achieving statistical significance (Figure 4D-E,G-H; supplemental Figure 3B). Only partial reductions in Fib-γ cross-linking were observed even when adjusted to Fibβ peak intensity, a strategy to account for variation in overall fibrin(ogen) deposition in each liver sample (supplemental Figure 4B). Similarly, only a partial reduction in cross-linked Fibγ (∼120 kDa) was observed using standard immunoblotting (supplemental Figure 3B). The results indicate a variable contribution of thrombin-catalyzed fibrin polymerization to hepatic fibrin(ogen) deposition after APAP overdose and that FXIII-dependent fibrin(ogen) cross-linking in the APAP-injured liver can occur without thrombin-mediated fibrin polymerization.
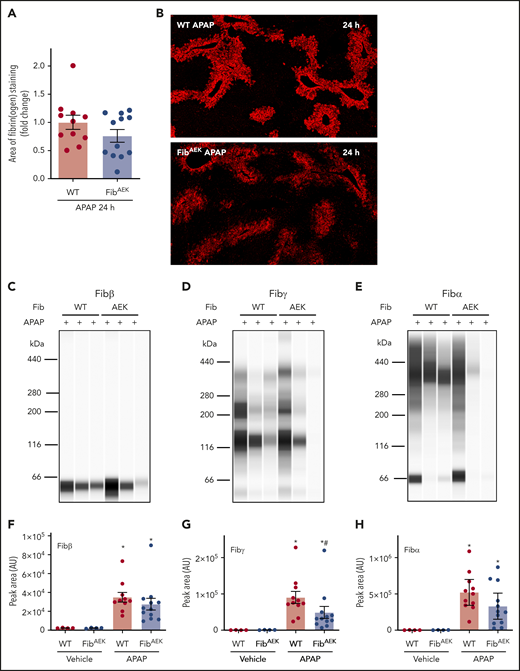
Role of thrombin-driven fibrin polymer formation in hepatic fibrin(ogen) cross-linking after APAP overdose. Mice expressing mutant fibrinogenAEK (FibAEK mice) and WT mice were treated intraperitoneally with saline vehicle or 300 mg/kg APAP. Livers were collected 24 hours after APAP challenge. (A) Fibrin(ogen) labeling in frozen liver sections was quantified (n = 11-12 mice/group). (B) Representative photomicrographs of fibrinogen-labeled frozen sections (×5 virtual magnification). Fibrin(ogen) levels were measured in enriched insoluble liver extracts using capillary-based western blotting (Wes). (C-E) Representative digital capillary images show fibrin(ogen) detected and quantified (F-H) by rabbit polyclonal antibodies selective for fibrin(ogen) Aα, Bβ, and γ chains. n = 4 mice for vehicle-treated groups and 11 to 12 mice for each APAP-challenged group (F-H). Data expressed as mean ± standard error of the mean. *Significantly (P < .05) different from vehicle-treated mice of the same genotype. #Significantly (P < .05) different from APAP-challenged WT mice.
Interestingly, the absence of thrombin-catalyzed fibrin polymer formation modestly increased APAP-induced liver injury in FibAEK mice compared with WT mice 24 hours after APAP challenge. Plasma ALT levels were significantly elevated in APAP-challenged FibAEK mice compared with WT mice, and centrilobular necrosis and congestion and hemorrhage tended to be higher in APAP-challenged FibAEK mice (Figure 5A-C). Thus, although thrombin-catalyzed fibrin polymerization drives only a fraction of fibrin(ogen) deposition in the injured liver, this pool of traditional fibrin polymer may contribute to the hepatoprotective response after APAP overdose.
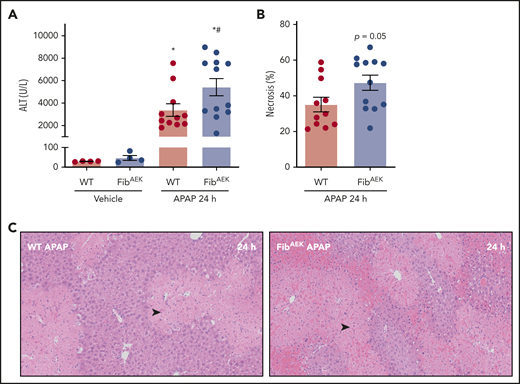
Role of thrombin-driven fibrin polymer formation in liver injury after APAP overdose. Mice expressing mutant fibrinogenAEK (FibAEK mice) and WT mice were treated intraperitoneally with saline vehicle of 300 mg/kg APAP and liver and citrated plasma were collected 24 hours after challenge. (A) Plasma ALT activity was determined using commercial reagents (n = 4 mice/group for vehicle treated and n = 11-12 mice/group for APAP challenged). (B) Area of centrilobular necrosis was quantified (n = 11-12 mice/group). (C) Representative photomicrographs of H&E-stained liver sections (necrosis denoted by black arrowhead). Data expressed as mean ± standard error of the mean. *Significantly (P < .05) different from vehicle-treated mice of the same genotype at 24 hours. #Significantly (P < .05) different from APAP-challenged WT mice.
Fibrin(ogen) is oxidatively modified in the APAP-injured liver
Fibrin(ogen) cross-linking occurred in the injured liver without a need for thrombin-mediated polymerization. We therefore sought to identify a potential mechanism that may permit fibrin(ogen) in the APAP-injured liver to undergo cross-linking in the absence of thrombin-mediated polymerization. We hypothesized that the necrotic liver microenvironment provides a mechanism whereby fibrin(ogen) cross-linking occurs even if fibrin polymerization fails. Previous studies indicated that local production of high levels of reactive nitrogen species (eg, PN, ONOO-) is a conspicuous feature of APAP hepatotoxicity.17 Notably, prior studies have suggested that PN inhibits fibrin polymerization.14,18 We sought to determine (1) whether fibrin(ogen) deposited in the injured liver is modified by PN and (2) the effect of PN on fibrin polymerization and cross-linking. As anticipated, fibrin(ogen) deposition was evident in whole liver homogenates, both 6 and 24 hours after APAP challenge (Figure 6A). Interestingly, fibrin(ogen) was immunoprecipitated from the APAP-injured liver using a monoclonal anti-nitrotyrosine antibody (Figure 6B), suggesting that fibrin(ogen) is indeed modified by ONOO- in the APAP-injured liver.
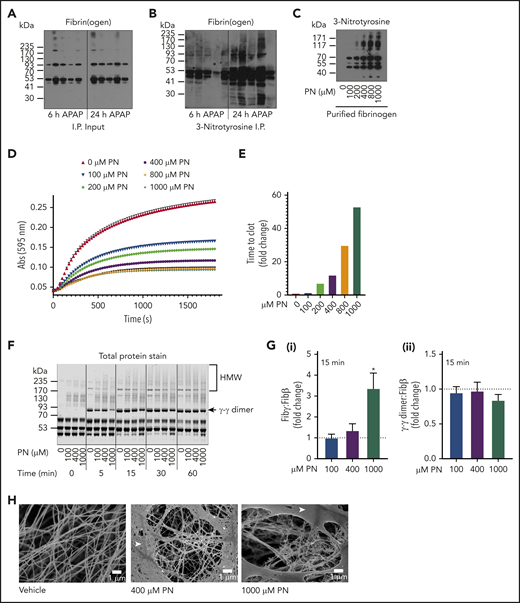
Detection of PN-modified fibrin(ogen) in the APAP-injured liver and effect of PN modification on fibrin polymerization and cross-linking. WT mice were challenged with 300 mg/kg APAP, and livers were collected 6 or 24 hours later to prepare homogenates (n = 5 mice/group). (A) Fibrin(ogen) detected by western blot using a rabbit anti-fibrinogen polyclonal antibody in whole-liver homogenates. (B-C) Immunoprecipitation of 3-NT adducted proteins from whole-liver homogenates and detection of fibrin(ogen) by western blot using a rabbit anti-fibrinogen polyclonal antibody. (B) 3-NT adducts in purified human fibrinogen modified by PN detected by western blotting (C). (D-E) Polymerization of purified human fibrinogen after PN modification was measured by turbidity (D) and modified Clauss assay (E). (F) Fibrin(ogen) cross-linking was assessed in clots prepared from vehicle- or PN-modified human fibrinogen incubated with thrombin for the indicated time points. (G) Human fibrinogen was reacted with the indicated concentration of PN in 3 independent reactions and then incubated with thrombin (0.25 U/mL) for 15 minutes. Quantitation of Fibγ (i) and γ-γ dimer (ii) in a Coomassie-stained gel (as shown in supplemental Figure 5) is expressed as a ratio to Fibβ compared with vehicle-modified fibrinogen (expressed as dashed line set at 1). *P < .05 compared with vehicle-modified fibrinogen. (H) Human fibrinogen was reacted with the indicated concentration of PN and observed using SEM. Fibers are visible beneath a fibrin film (white arrowhead). Representative photomicrographs are shown (×10 000 magnification).
Having identified that fibrin(ogen) is modified by PN in the APAP-injured liver, we next examined the effect of PN on fibrin polymerization by thrombin and cross-linking by FXIIIa. PN treatment increased 3-NT modification of human fibrinogen in a concentration-dependent manner (Figure 6C). In agreement with prior studies,14 pretreatment of fibrinogen with increasing concentrations of PN reduced fibrin clot turbidity in a concentration-dependent manner, indicated by decreased maximum absorbance at 595 nm (Figure 6D). PN also increased the time required to stable clot formation in response to thrombin treatment (Figure 6E). Interestingly, impaired polymerization did not prevent FXIII-mediated cross-linking, as indicated by similar levels of γ-γ dimer (∼93 kDa) and HMW cross-linked fibrin(ogen) in fibrin clots prepared from PN-modified fibrin(ogen) subjected to reducing conditions (Figure 6F). Interestingly, PN appeared to delay but not prevent fibrin cross-linking (Figure 6F). With the exception of the highest concentration of PN, consumption of Fibγ monomer and formation of γ-γ dimer were largely complete within 15 minutes of thrombin addition (Figure 6G; supplemental Figure 5). Interestingly, residual Fibγ (∼46 kDa) was present in fibrinogen pretreated with 1000 µM PN, although this was not connected to a delayed appearance of γ-γ dimer in all 3 experimental replicates, which are presented in supplemental Figure 5. Using SEM, we observed that untreated fibrin(ogen) formed a prototypical fibrin network that could be seen beneath a dense fibrin film (white arrowheads; Figure 6H; supplemental Figure 6). Furthermore, we observed a concentration-dependent effect of PN on fibrin network formation, including obvious disorganization of the fibrin networks with fewer fibrils and more open space between the fibers, suggesting a defect in fibrin polymerization (Figure 6H), an effect most pronounced at the highest PN concentration (supplemental Figure 6A). In contrast to control fibrin, PN-modified fibrin was very heterogenous, wherein some locations completely lacked an obvious fibrin network below the film (supplemental Figure 6B). Taken together, these results indicate that PN disturbs fibrin polymerization, suggested by changes in turbidity, clotting time, and clot structure, whereas fibrin(ogen) cross-linking is minimally affected.
Discussion
Previous studies have suggested that thrombin-induced fibrin polymerization is required for efficient fibrin cross-linking by FXIIIa.3 Prior studies provide strong evidence in support of this conclusion using ex vivo cross-linked fibrin clot formation, biochemical assays, and various experimental settings of intravascular thrombosis.4,6,19-21 It has also been demonstrated in vitro that under certain conditions, FXIIIa can cross-link fibrinogen, although at a much slower rate. However, there has been no experimental evidence to date suggesting that FXIII can cross-link fibrinogen independently of fibrin polymer formation in vivo. In the current study, we document evidence of a pathologic condition in which fibrin polymer formation is not required for fibrin(ogen) cross-linking by FXIII. Specifically, this occurred during acute liver damage, wherein large amounts of fibrin(ogen) accumulate within areas of hepatic necrosis. Cross-linked fibrin(ogen) accumulated in the APAP-injured liver even in mice expressing mutated fibrinogen fully resistant to thrombin-mediated fibrin polymerization. The formation of intrahepatic cross-linked HMW fibrin(ogen) species was found to be largely FXIII-dependent in APAP-challenged mice. The interpretation of these combined observations is that acute hepatocellular necrosis provides a pathologic microenvironment allowing cross-linking of fibrin(ogen) by FXIII independent of thrombin-mediated polymerization.
Prior studies indicated that APAP overdose is associated with rapid coagulation cascade activation.7 The predominant assumption has been that thrombin activity drives fibrin(ogen) accumulation in the acutely injured liver. Tissue factor (TF)-initiated coagulation activation, indicated by increased plasma thrombin–antithrombin complexes, peaks within a few hours (ie, 1-3) of APAP overdose.7,10,16 In contrast, hepatic fibrin(ogen) deposition in the injured liver more closely parallels the severity of necrosis, which peaks 18 to 24 hours after APAP overdose.7,10 Indeed, global TF deficiency did not affect peak hepatic fibrin(ogen) deposition in APAP-challenged mice.7 Liver-specific TF deficiency also did not reduce hepatic fibrin(ogen) deposition 24 hours after APAP overdose (data not shown). Our studies using APAP-challenged FibAEK mice resolve these findings by demonstrating a functional disconnect between fibrin(ogen) deposition and coagulation activity during the progression of liver damage. Notably, fibrin(ogen) accumulated in livers of both WT and FibAEK mice after acute challenge with carbon tetrachloride, another hepatotoxicant that causes centrilobular hepatic necrosis (data not shown). In contrast, fibrin polymer formation was absent in the livers of FibAEK mice after a liver biopsy injury,11 suggesting that the mechanisms driving fibrin(ogen) accumulation in the injured liver are context dependent.
Although a variety of experimental settings of liver injury are often united under the header of “sterile liver injury,” there are clear differences in the timing and etiology of tissue injury. Unlike the brief and well-defined liver biopsy injury,11 APAP overdose is associated with progressive hepatocellular injury driven by a well-ordered series of molecular events and exacerbated by inflammatory tissue injury and oxidative stress.17,22 Our results suggest that a component of this mechanism enables (1) fibrin(ogen) to deposit in the injured liver independent of coagulation activity and (2) undergo FXIII-dependent cross-linking independent of fibrin polymerization. Traditional cross-linked fibrin polymer deposition in APAP-challenged mice may parallel coagulation activation, with an alternate mechanism favored as liver necrosis becomes severe. Indeed, although peak fibrin(ogen) deposition 24 hours after APAP overdose is unaffected in mice expressing low tissue factor, early fibrin(ogen) deposition (6 hours after APAP challenge) was significantly reduced in these mice.7 We proposed that robust oxidative stress evident in the APAP-injured liver may form a mechanism impairing local fibrin polymerization, which when combined with the extended duration of extravascular fibrin(ogen) deposition, may permit direct FXIII cross-linking of fibrinogen monomer. Hepatic oxidative stress is a well-recognized and mechanistically relevant feature of APAP-induced liver necrosis, with increased levels of an assortment of reactive oxygen and nitrogen species.17 Oxidative modification of fibrinogen has been shown to delay fibrin polymerization and to alter polymer thickness.14,23 Building on prior studies, we found that ONOO-impairs thrombin-catalyzed fibrin polymerization, but only delays FXIII-dependent fibrin(ogen) cross-linking. Although additional mechanisms should not be excluded, our studies document 3-NT–modified fibrinogen in the APAP-injured liver, forming a plausible basis for FXIII-dependent fibrin(ogen) cross-linking independent of proper fibrin polymer formation.
Our studies identified HMW Fibα and Fibγ complexes in the APAP-injured liver. These HMW fibrin(ogen) complexes appeared at distinct molecular weights depending on sample preparation and detection method used, even for Fibγ, which is not traditionally thought to undergo cross-linking other than dimerization. In the necrotic microenvironment of the liver, it is possible that fibrin(ogen) may not only be cross-linked to itself but also to other proteins. Future studies should seek to identify the peptide composition of these fibrin(ogen) complexes. Furthermore, it is possible that, in addition to FXIII, other transglutaminases such as tissue transglutaminase (TG2) determine the final composition of cross-linked fibrin(ogen) complexes in the APAP-injured liver. TG2 is known to cross-link fibrin(ogen), even in the absence of thrombin-mediated polymerization, to form Fibα-γ cross-links,24 and our previous studies suggest that TG2 contributes to fibrin(ogen) deposition in experimental chronic liver injury.13 In our studies, hepatic TG2 expression was unaltered by FXIII deficiency (supplemental Figure 7), suggesting the possibility that it could drive residual fibrin(ogen) cross-linking in these mice. In addition to forming unique Fibα-γ cross-links, TG2 cross-linking of fibrin(ogen) has been shown to increase resistance of fibrin(ogen) to lysis.25 Future studies using proteomic approaches should seek to identify the precise peptide composition of these HMW complexes, as such studies could uncover novel functions of not only FXIII but other transglutaminases in the APAP-injured liver.
APAP metabolism causes direct hepatocyte injury, and this is exacerbated by numerous mechanisms. Several studies have shown that anticoagulant drugs reduce early APAP hepatotoxicity. Ganey et al7 found that treatment of mice with heparin significantly reduced early APAP hepatotoxicity (ie, 6 hours after APAP challenge). Subsequent studies have validated this finding using the direct thrombin inhibitors lepirudin and dabigatran.8,16 The mechanism whereby thrombin drives early hepatotoxicity appears to involve activation of protease-activated receptors (PARs; eg, PAR-1 and PAR-4).7,16 Importantly, our studies in FibAEK mice indicate that fibrin polymerization contributes to the hepatoprotective effects of fibrin(ogen) in the APAP-injured liver. Indeed, APAP-induced hepatocellular necrosis was modestly elevated 24 hours after APAP challenge in FibAEK mice compared with WT mice. This result is important because it establishes clear dichotomous roles for thrombin in APAP hepatotoxicity. Specifically, early injury is exacerbated by PAR activation, whereas thrombin drives hepatoprotective pathways by promoting fibrin polymerization. Indeed, this provides a much-needed explanation for prior studies showing that initial beneficial effects of anticoagulation are lost as injury progresses to repair.8 Dual treatment with PAR-1 or PAR-4 antagonists could plausibly inhibit both liver-expressed PARs and perhaps reduce the pathologic effects of platelets in this setting,16 with a major limitation to translation being concern about tipping the delicately rebalanced hemostasis in these patients.26 Another novel possibility alluded to by our results is to promote formation of hepatoprotective fibrin polymer in the injured liver by inhibition of its oxidative modification. This may uncover a novel mechanism whereby molecules like PN contribute to liver damage.17
Prior studies suggest that the hepatoprotective effects of fibrin(ogen) are conferred, at least in part, by the interaction between fibrin(ogen) and leukocyte β2 integrins. Liver necrosis was increased and repair responses decreased in APAP-challenged Fibγ390-396A mice, which express normal levels of a mutant fibrinogen that cannot engage β2 integrins.8 Several β2 integrin expressing cells limit APAP hepatotoxicity and confer prorepair functions in the APAP-injured liver.27 Importantly, liver injury in APAP-challenged FibAEK mice mirrored some histopathologic features of APAP-challenged Fibγ390-396A mice. Although our initial studies lacked statistical power to detect a difference in injury markers, analyzing results from multiple studies revealed a modest increase in necrosis (P = .05) and regular evidence of congestion/hemorrhage in APAP-challenged FibAEK mice. In contrast, FXIII deficiency had no significant effect on APAP-induced liver necrosis, despite a dramatic reduction in fibrin(ogen) cross-linking. Collectively, these studies suggest that fibrin polymer formation, but not cross-linking, is essential for hepatoprotective fibrin(ogen)–β2 interactions. This also makes it unlikely that the phenotype of APAP-challenged Fibγ390-396A mice is solely a consequence of delayed FXIII-dependent cross-linking.28 Indeed, it is also possible that the protective effects of fibrin polymer involve multiple mechanisms, among them supporting the fibrin(ogen)–β2 interaction to drive macrophage prorepair function. Moreover, the role of oxidatively modified fibrin(ogen) deposited in the injured liver, either as a dysfunctional remnant or unique effector molecule, deserves further exploration.
In summary, we detail the first example of an in vivo experimental setting wherein FXIII-dependent fibrin(ogen) cross-linking proceeds even in the absence of effective fibrin polymerization. The results suggest oxidative modification of fibrinogen common to robust chemical-induced liver injury enables this pathway. Interestingly, generation of reactive oxygen species and formation of 3-NT adducts is evident in liver injury driven by multiple challenges, including toxicants,29 alcohol,30 and high-fat diet.31 Future studies should investigate the contribution of reactive oxygen species generation to fibrin(ogen) deposition in the injured liver in these settings. A complementary and equally novel observation is that, although fibrin(ogen) accumulation in the injured liver does not depend on coagulation, thrombin-catalyzed polymerization of fibrin(ogen) inhibits liver damage. Collectively, these results highlight the discovery and in vivo relevance of a nontraditional fibrinogen deposition and cross-linking mechanism relevant to liver injury.
For original data, please contact luyendyk@msu.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Carol Flegler in the MSU Center for Advanced Microscopy for expertise and dedicated assistance with SEM analysis.
This research was supported by grants from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK105099 and R01 DK120289; J.P.L.), (R01DK112778; M.J.F.), and (F32 DK121423; L.G.P.); a research grant from the European Hematology Association (D.J.G.); a John A. Penner endowed research assistantship to K.S.B.; and support from the US Department of Agriculture National Institute of Food and Agriculture (J.P.L.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: L.G.P., A.K.K., A.P., D.J.G., K.S.B., H.M.C.-F., M.J.F., and J.P.L. designed the studies; L.G.P., A.K.K., A.P., D.J.G., K.S.B., and H.M.C.-F. performed experiments and collected data; M.J.F. contributed vital mouse lines for the studies; L.G.P., A.K.K., D.J.G., M.J.F., and J.P.L. analyzed and interpreted the data; L.G.P., A.K.K., and J.P.L. drafted the manuscript; and all authors reviewed and approved the final version of the manuscript for submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: James P. Luyendyk, Department of Pathobiology and Diagnostic Investigation, Michigan State University, 1129 Farm Lane, Room 253, East Lansing, MI 48824; e-mail: luyendyk@msu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal