

Key Points
CAR22 therapy induced complete remission in 3 patients with LBCL who had relapsed after CAR19 therapy and caused no severe toxicities.

Abstract
The prognosis of patients with large B-cell lymphoma (LBCL) that progresses after treatment with chimeric antigen receptor (CAR) T-cell therapy targeting CD19 (CAR19) is poor. We report on the first 3 consecutive patients with autologous CAR19-refractory LBCL who were treated with a single infusion of autologous 1 × 106 CAR+ T cells per kilogram targeting CD22 (CAR22) as part of a phase 1 dose-escalation study. CAR22 therapy was relatively well tolerated, without any observed nonhematologic adverse events higher than grade 2. After infusion, all 3 patients achieved complete remission, with all responses continuing at the time of last follow-up (mean, 7.8 months; range, 6-9.3). Circulating CAR22 cells demonstrated robust expansion (peak range, 85.4-350 cells per microliter), and persisted beyond 3 months in all patients with continued radiographic responses and corresponding decreases in circulating tumor DNA beyond 6 months after infusion. Further accrual at a higher dose level in this phase 1 dose-escalation study is ongoing and will explore the role of this therapy in patients in whom prior CAR T-cell therapies have failed. This trial is registered on clinicaltrials.gov as #NCT04088890.
Introduction
Outcomes of patients with large B-cell lymphoma (LBCL) that has relapsed after or is refractory (R/R) to chimeric antigen receptor T-cell therapy targeting CD19 (CAR19) remain poor, with <25% responding to subsequent therapies, and a median overall survival (OS) of 3.6 months.1 Targeting of alternative antigens represents an important therapeutic strategy for patients who relapse after CAR19, including those with CD19 loss or downregulation.1,2 CD22 is expressed on LBCL cells at levels similar to those found on nonmalignant B-cells3,4 and is an effective target for CAR T-cell therapy (CAR22) in children with R/R acute lymphoblastic leukemia.5,6 No CD22-directed therapy is currently approved for use in LBCL, and only 2 cases of CD22-directed CAR T-cell therapy against LBCL have been described.7,8 We report the safety and interim efficacy of the initial dosing cohort of a phase 1 dose-escalation study of CD22.BB.z-CAR in adults with LBCL that relapsed after CAR19.
Study design
Eligible patients had to have received at least 2 prior lines of therapy for LBCL, with measurable disease that demonstrated any level of CD22 expression. For patients who received prior CAR T-cell therapy, CAR+ cells had to represent <5% of peripheral blood CD3+ lymphocytes. Lymphodepletion with fludarabine 30 mg/m2 and cyclophosphamide 500 mg/m2 preceded infusion of 1 × 106 CD22.BB.z-CAR+ cells per kilogram (Figure 1A). CAR T-cell–related toxicity was graded according to American Society for Transplantation and Cellular Therapy consensus criteria9 and was managed via institutional protocol. Common Terminology Criteria for Adverse Events (v5.0) and Lugano criteria10 were used to grade adverse events and responses, respectively. The trial protocol received institutional review board approval. Patients provided informed consent in accordance with the Declaration of Helsinki. Products were manufactured in the automated, closed-system Miltenyi CliniMACS Prodigy device. CD4/CD8 T-cell–enriched peripheral blood mononuclear cells were transduced with a lentiviral vector containing a single-cistron–encoded CD22.BB.z-CAR cell comprising a fully humanized CD22 scFv (m971), a CD8 transmembrane domain, a 4-1BB costimulatory domain, and a CD3ζ activation domain (supplemental Figure 1, available on the Blood Web site). Products were phenotypically similar and were predominantly CD4+ T cells (supplemental Figure 2). Details regarding patient eligibility, product manufacturing, toxicity management, and in vivo correlative studies are included in the supplemental Methods.
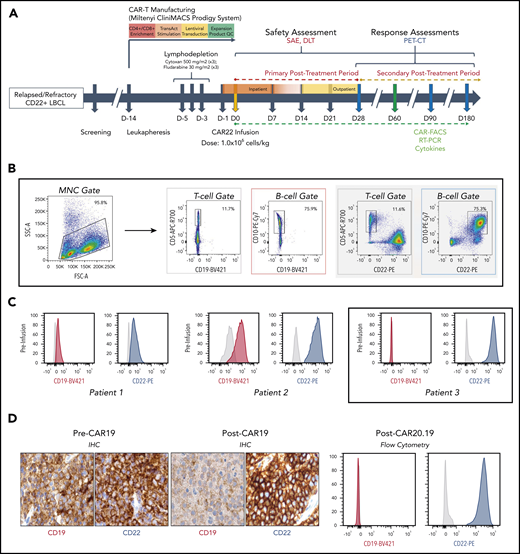
Protocol schema and assessment of target antigen expression CAR22 therapy. (A) Overview of the trial design, including enrollment, manufacturing of autologous CD22-directed CAR T cells, administration of the therapy, and follow-up monitoring. Patients had safety assessments performed and blood samples drawn at each arrow after infusion, for assessment of correlative biomarkers, and underwent clinical and radiographic response assessment at each blue arrow. The green arrow indicates the time period during which CAR-FACS, RT-PCR, and cytokines were collected at all specified time points. (B) Gating strategy and (C) flow cytometry histograms of CD19 and CD22 expression for each patient before CAR22 therapy. After CAR19 therapy, P1 demonstrated preserved CD19 expression, whereas P2 had heterogeneous and downregulated CD19 expression. In all 3 patients, CD22 expression was preserved at high levels. (D) Serial biopsy specimens of P3 showing CD19 downregulation after CAR19 therapy, then complete loss of CD19 expression after CAR20.19 therapy. Original IHC image magnification ×40. Anti-CD19 antibody clone BT51E (murine monoclonal, Leica #PA0843). Anti-CD22 antibody clone FPC1 (murine monoclonal, Leica #PA0249). CAR19, anti-CD19 CAR T-cell therapy; CAR20.19, anti-CD19, anti-CD20 tandem CAR T-cell therapy; CAR-FACS, high dimensional flow cytometry immunophenotyping of peripheral blood, including identification of CAR+ cells; DLT, dose-limiting toxicity; FSC-A, forward scatter area; IHC, immunohistochemistry; MNC, mononuclear cell; PET-CT, composite positron emission tomographic-computed tomographic imaging; qRT-PCR, quantitative reverse transcriptase-polymerase chain reaction for identification of CAR transgene copies in peripheral blood; SAE, severe adverse event; SSC-A, side scatter area.
Protocol schema and assessment of target antigen expression CAR22 therapy. (A) Overview of the trial design, including enrollment, manufacturing of autologous CD22-directed CAR T cells, administration of the therapy, and follow-up monitoring. Patients had safety assessments performed and blood samples drawn at each arrow after infusion, for assessment of correlative biomarkers, and underwent clinical and radiographic response assessment at each blue arrow. The green arrow indicates the time period during which CAR-FACS, RT-PCR, and cytokines were collected at all specified time points. (B) Gating strategy and (C) flow cytometry histograms of CD19 and CD22 expression for each patient before CAR22 therapy. After CAR19 therapy, P1 demonstrated preserved CD19 expression, whereas P2 had heterogeneous and downregulated CD19 expression. In all 3 patients, CD22 expression was preserved at high levels. (D) Serial biopsy specimens of P3 showing CD19 downregulation after CAR19 therapy, then complete loss of CD19 expression after CAR20.19 therapy. Original IHC image magnification ×40. Anti-CD19 antibody clone BT51E (murine monoclonal, Leica #PA0843). Anti-CD22 antibody clone FPC1 (murine monoclonal, Leica #PA0249). CAR19, anti-CD19 CAR T-cell therapy; CAR20.19, anti-CD19, anti-CD20 tandem CAR T-cell therapy; CAR-FACS, high dimensional flow cytometry immunophenotyping of peripheral blood, including identification of CAR+ cells; DLT, dose-limiting toxicity; FSC-A, forward scatter area; IHC, immunohistochemistry; MNC, mononuclear cell; PET-CT, composite positron emission tomographic-computed tomographic imaging; qRT-PCR, quantitative reverse transcriptase-polymerase chain reaction for identification of CAR transgene copies in peripheral blood; SAE, severe adverse event; SSC-A, side scatter area.
Results and discussion
Patients
Three patients with R/R LBCL, in whom the disease had progressed after multiple treatments, including CAR19 therapy, were enrolled and treated as the first 3 consecutive patients in this clinical study of CAR22. Patient and disease characteristics at the time of enrollment are shown in supplemental Table 1. None had achieved a durable remission with any prior treatment (supplemental Table 2), including CAR19, after which patients 1 and 2 (P1 and P2) had achieved only a partial response (PR). All patients had progressive disease by 3 months after CAR19 infusion (supplemental Figure 3A). Patient 3 (P3) had progressed after CAR19 and bispecific CD19- and CD20-targeted CAR (CAR20.19; supplemental Figure 3B).11 All patients had biopsy-proven expression of surface CD22 detected by flow cytometric cell sorting (FACS) and immunohistochemistry, whereas CD19 expression was downregulated or lost in 2 patients (P2 and P3; Figure 1C; supplemental Figure 4). P3 had surface CD19 downregulation after CAR19, followed by complete loss of CD19 after CAR20.19, whereas CD22 remained preserved throughout (Figure 1D).
Safety and adverse event profile
No nonhematologic treatment–emergent adverse events of grade ≥3 occurred (supplemental Table 3). Grade 1 cytokine release syndrome (CRS) occurred in all patients within 24 hours after CAR22 infusion. P1 and P3 developed grade 2 CRS on days 11 and 7 after infusion, respectively; each received tocilizumab and dexamethasone with subsequent resolution. No patient demonstrated evidence of immune effector cell–associated neurotoxicity syndrome. All patients developed grade ≥3 neutropenia, thrombocytopenia, and anemia after lymphodepletion chemotherapy and CAR22 infusion, and recovery to grade ≤2 cytopenias did not occur until nearly 4 months after infusion (supplemental Figure 5).
Clinical and molecular response
All patients achieved a complete response (CR) as the best response after CAR22. Progressive reduction in the size of all index lesions was observed over the course of follow-up (Figure 2A; supplemental Table 3). Both patients with high tumor burden (P1 and P2) had initial PR at day 28, which subsequently improved to CR at 6 and 3 months, respectively (Figure 2B). At a mean follow-up of 7.8 months (range, 6-9.3), all patients remained in CR. Similarly, circulating tumor DNA (ctDNA) was undetectable (P1 and P3) or reduced by 99% (P2) from baseline at 6 months after infusion, demonstrating persistent CAR22 activity (Figure 2C).
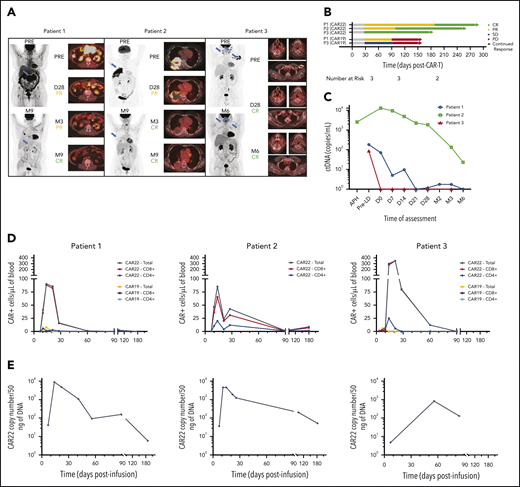
CAR22 cells expand and persist in vivo and induce complete clinical responses associated with reductions in ctDNA. (A) Maximum-intensity projections (MIPs) and PET-CT composite cross-sectional imaging for primary index lesions at specified assessment time points after infusion of CAR22 therapy. Two-dimensional MIP images are shown on the left of each panel, with blue arrows indicating index lesions shown in the cross-sectional imaging to the right. Response classifications at each time point are according to Lugano criteria. (B) Swimmer plot demonstrating late conversion of PR to CR in P1 and P2, as well as durability compared with prior CAR19 responses of P1 and P3. (C) ctDNA levels were consistently reduced after CAR22 therapy. CAR+ T-cell expansion and persistence as measured by flow cytometry (D) and quantitative PCR (E) measurements in all patients. CD8+ T cells were the predominant subset expanding in vivo, and CAR+ cells remained detectable in circulation up to 6 months. APH, apheresis; D28, day 28 after infusion; M3, day 90 after infusion; M6, day 180 after infusion; M9, day 270 after infusion; PD, progressive disease; PRE, preinfusion/baseline assessment; Pre-LD, pre-lymphodepletion chemotherapy; SD, stable disease.
CAR22 cells expand and persist in vivo and induce complete clinical responses associated with reductions in ctDNA. (A) Maximum-intensity projections (MIPs) and PET-CT composite cross-sectional imaging for primary index lesions at specified assessment time points after infusion of CAR22 therapy. Two-dimensional MIP images are shown on the left of each panel, with blue arrows indicating index lesions shown in the cross-sectional imaging to the right. Response classifications at each time point are according to Lugano criteria. (B) Swimmer plot demonstrating late conversion of PR to CR in P1 and P2, as well as durability compared with prior CAR19 responses of P1 and P3. (C) ctDNA levels were consistently reduced after CAR22 therapy. CAR+ T-cell expansion and persistence as measured by flow cytometry (D) and quantitative PCR (E) measurements in all patients. CD8+ T cells were the predominant subset expanding in vivo, and CAR+ cells remained detectable in circulation up to 6 months. APH, apheresis; D28, day 28 after infusion; M3, day 90 after infusion; M6, day 180 after infusion; M9, day 270 after infusion; PD, progressive disease; PRE, preinfusion/baseline assessment; Pre-LD, pre-lymphodepletion chemotherapy; SD, stable disease.
CAR22 persistence and inflammatory response
Serial flow cytometric evaluation of peripheral blood (CAR-FACS) demonstrated a 100- to 400-fold expansion, with peak CAR+ T-cell levels occurring at ∼14 days. Circulating CAR+ T cells persisted for at least 3 months in all patients and were detectable by quantitative polymerase chain reaction (PCR) at the time of last assessment up to 6 months (Figure 2D-E). P1 and P3, who had received prior CAR19 therapy at our institution, experienced an 11.7- and 55.9-fold higher peak CAR+ T-cell level with CAR22 vs CAR19 therapy. Plasma levels of 48 cytokines were assessed over the first 28 days after infusion (supplemental Figure 7). In both patients who developed grade 2 CRS (P1 and P3), interleukin-6 and interleukin-1 receptor antagonist levels reached 7.7- to 12.1-fold and 3.7- to 4.6-fold higher concentrations before the event.
Conclusions and future directions
We report the first experience of autologous CAR T cells targeting CD22 for R/R LBCL and demonstrate ongoing CR in the first 3 consecutive patients treated. CAR22 induced clinical and molecular CRs despite prior failure of autologous CAR19 therapy.
Currently, data regarding retreatment with CAR T-cell therapies are limited. In patients with LBCL who relapse after CAR19, reinfusion with an identical or derivative CAR19 product has not produced a durable response.12 This report provides additional evidence that targeting CD22 can mediate meaningful clinical activity in LBCL, even in the post-CAR19 relapse setting.13
CD22 antigen density has been shown to be an important factor that modulates the persistence and efficacy of CAR22+ cells in B-acute lymphoblastic leukemia.6,14 In 2 of our patients, CD22 expression was maintained at high levels, despite downregulation or loss of CD19 in response to the selective pressure of CAR19 therapy. The current ongoing clinical study will help inform the effect of CD22 antigen density on the response to CAR22 therapy for LBCL.
In all patients, CAR22 demonstrated robust expansion and persistence, similar to levels reported in CAR T-cell–naive patients who received 41BB-based CAR19.15,16 The corresponding rapid and pleiotropic changes in infusion levels of inflammatory and effector cytokines mimicked patterns that have been shown to be associated with improved antitumor activity.17,18 The absence of severe immune effector cell–associated neurotoxicity syndrome, or CRS, is noteworthy, given that analogous patterns of CAR+ T-cell expansion and inflammatory response are often associated with these toxicities in patients who undergo CAR19 therapy.19-21
The use of CD4/CD8-enriched apheresis material with the CliniMACS Prodigy platform resulted in products with a predominance of CD4+ T cells in all patients, in line with previous reports on a CD22.BB.z-CAR.6 However, in vivo CAR+ T-cell expansion was largely CD8+ T cells. Despite a minority of T cells in the product that exhibited an early memory phenotype (both central memory and naïve memory), which has been associated with greater persistence in vivo, we observed detectable CAR+ cells beyond 6 months after infusion.22,23 Studies are ongoing to examine the clonal dynamics of CAR22 in vivo and better understand the contribution of these populations to response and toxicity.
In summary, we provide initial evidence that demonstrates the safety and antitumor activity of CAR T-cell therapy targeting CD22 in patients with R/R LBCL, establishing CD22 as a potential target for CAR T-cell therapy in LBCL. Further accrual at a higher dose level in this phase 1 dose-escalation study is ongoing and will explore the role of this therapy in patients in whom prior CAR T-cell therapies have failed.
Deidentified individual participant data that underlie the reported results will be made available for approved use by the study authors. Proposals for access should be sent to lmuffly@stanford.edu. Complete trial cohort-level data will be published on clinicaltrials.gov at the conclusion of the trial.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the staff and faculty of the Stanford University Blood and Marrow Transplantation Program and the Stanford Center for Cancer Cell Therapy for their tireless work in caring for the patients involved in this study.
This work was supported by National Institutes of Health (NIH), National Cancer Institute (NCI) grants 2P01CA049605-29A1 (C.L.M., R.S.N., D.B.M.), 5P30CA124435 (C.L.M.), and K08CA248968 (M.J.F.) and by the Virginia and D. K. Ludwig Fund for Cancer Research. C.L.M is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: L.M., M.J.F., C.L.M., and D.B.M. designed the study; J.H.B., L.M., and M.J.F. interpreted the data; J.H.B., S.P., J.T., L.M., and M.J.F. analyzed the data and created the figures; J.H.B., L.M., M.J.F., D.B.M., and C.L.M. drafted the manuscript; and all authors contributed to data collection, writing and revision of the manuscript, and approval of the final version.
Conflict-of-interest disclosure: P.S. has received research support from Kite Pharma-Gilead. A.R.R. has received research support from Pharmacyclics/AbbVie, served on 1-time ad hoc scientific advisory boards of Nohla Therapeutics and Kaleido, and was an expert witness for the U.S. Department of Justice. His brother works for Johnson & Johnson. C.L.M. has consulted for Lyell, Neoimmune Tech, Nektar, and Apricity; has received royalties from NIH and Juno Therapeutics for CD22-CAR; holds equity in Lyell and Apricity; and has received research support from Lyell. D.B.M. has consulted for Kite Pharma-Gilead, Juno Therapeutics-Celgene, Novartis, Janssen, and Pharmacyclics and has received research support from Kite Pharma-Gilead, Allogene, Pharmacyclics, Miltenyi Biotec, and Adaptive Biotechnologies. S.S. has consulted for Janssen. C.M., A.J., and I.K. are full-time employees of Adaptive Biotechnologies. S.A.F. has consulted for Lonza PerMed, Gradalis, Obsidian, and Samsara BioCapital; L.M. has received research support from Adaptive Biotechnologies and Servier Laboratories and has consulted for Amgen and Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Lori Muffly, Stanford University, 300 Pasteur Dr, H0144, Stanford, CA 94305; e-mail: lmuffly@stanford.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal