

Key Points
Tumor mutations in STK11, KRAS, CTNNB1, KEAP1, CDKN2B, MET, and SETD2 modulate the risk of cancer-associated thrombosis.
The presence of clonal hematopoiesis does not affect the risk of cancer-associated thrombosis.

Abstract
Venous thromboembolism (VTE) associated with cancer (CAT) is a well-described complication of cancer and a leading cause of death in patients with cancer. The purpose of this study was to assess potential associations of molecular signatures with CAT, including tumor-specific mutations and the presence of clonal hematopoiesis. We analyzed deep-coverage targeted DNA-sequencing data of >14 000 solid tumor samples using the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets platform to identify somatic alterations associated with VTE. End point was defined as the first instance of cancer-associated pulmonary embolism and/or proximal/distal lower extremity deep vein thrombosis. Cause-specific Cox proportional hazards regression was used, adjusting for pertinent clinical covariates. Of 11 695 evaluable individuals, 72% had metastatic disease at time of analysis. Tumor-specific mutations in KRAS (hazard ratio [HR], 1.34; 95% confidence interval (CI), 1.09-1.64; adjusted P = .08), STK11 (HR, 2.12; 95% CI, 1.55-2.89; adjusted P < .001), KEAP1 (HR, 1.84; 95% CI, 1.21-2.79; adjusted P = .07), CTNNB1 (HR, 1.73; 95% CI, 1.15-2.60; adjusted P = .09), CDKN2B (HR, 1.45; 95% CI, 1.13-1.85; adjusted P = .07), and MET (HR, 1.83; 95% CI, 1.15-2.92; adjusted P = .09) were associated with a significantly increased risk of CAT independent of tumor type. Mutations in SETD2 were associated with a decreased risk of CAT (HR, 0.35; 95% CI, 0.16-0.79; adjusted P = .09). The presence of clonal hematopoiesis was not associated with an increased VTE rate. This is the first large-scale analysis to elucidate tumor-specific genomic events associated with CAT. Somatic tumor mutations of STK11, KRAS, CTNNB1, KEAP1, CDKN2B, and MET were associated with an increased risk of VTE in patients with solid tumors. Further analysis is needed to validate these findings and identify additional molecular signatures unique to individual tumor types.
Introduction
Venous thromboembolism (VTE) is a frequent complication of cancer and a leading cause of cancer-related mortality.1,2 Clinical risk factors known to portend an increased risk of cancer-associated VTE (CAT) include body mass index, prior VTE events, inherited thrombophilias, blood cell counts, exposure to chemotherapy or recombinant erythropoietin, cancer stage, and the presence of underlying comorbid conditions, including infection.3-14 These factors form the basis of risk assessment models frequently used to identify patients most likely to benefit from anticoagulant prophylaxis, most commonly the Khorana score.3,15 However, despite improving CAT risk prediction, significant limitations remain with these models, and the optimal approach to pharmacologic prophylaxis remains unclear. Identification of additional risk factors to enhance current CAT prediction models might help to better personalize VTE prevention and improve clinical outcomes for patients with solid tumors.
The mechanisms driving the pathogenesis of CAT are likely complex and multifactorial, and they highlight the inextricably linked and highly dynamic interactions between the tumor, its microenvironment, and the hemostatic system. Known tumor-specific factors shown to promote venous thrombosis in cancer include: (1) overexpression and secretion of various procoagulant factors by the tumor, including tissue factor (TF) and TF-bearing microparticles; (2) activation of platelets and/or leukocytes by tumor-secreted factors, including proinflammatory cytokines; and/or (3) secondary effects of tumor cells on the surrounding vasculature and tissue microenvironment as a result of aberrant proangiogenic growth factor stimulation.16 Importantly, VTE rates vary significantly based on tumor subtype, and even according to histologic subtype within tumor types,17,18 suggesting that the individual mechanisms promoting thrombosis and the degree to which the coagulation system is perturbed are highly tumor specific.
In this manner, cancer cell genotype is increasingly recognized as an important factor for VTE development.19 Tumor-specific mutations in KRAS,20,21 ALK,22,23 and ROS124 have been associated with an increased risk of thromboembolism. Results for EGFR mutational status and VTE risk have been mixed, with only one study showing an increased risk and others showing no effect or even a decreased risk of VTE.20,25-28 Similarly, although many high-grade gliomas overexpress TF in hypoxic conditions, the presence of an IDH1 mutation seems to be protective against VTE in part due to reduced TF expression, further highlighting the influential role of tumor genotype in influencing CAT risk.29-32 Also, evidence from an animal model suggests that tumor-specific MET mutations might be associated with a hypercoagulable state.33 Overexpression of TF and other procoagulant factors along with enhanced neoangiogenesis via constitutive vascular endothelial growth factor stimulation are proposed mechanisms for the thrombophilic effect of oncogenic mutations.34 Whether the procoagulant state of these tumors is simply a byproduct of aberrant signaling or serves directly to further promote tumor growth remains unclear; regardless, these data underscore the urgent need to identify the full extent of molecular events that might contribute to cancer-related thrombosis.
The emerging entity of clonal hematopoiesis (CH) is also increasingly recognized as influencing thrombotic risk.35 CH has been found to occur in >10% to 20% of otherwise healthy individuals aged >70 years and in >25% of patients with solid tumor malignancies, and it is associated with adverse clinical outcomes.35,36 Importantly, studies have implicated CH in an increased rate of arterial thrombotic events, including myocardial infarction and stroke in otherwise healthy individuals.37 In one retrospective series, JAK2 V617F mutant CH was also found to be associated with an increased risk of VTE and early-onset atherothrombotic disease38 ; however, the role of CH influencing rates of VTE within the cancer patient population remains unknown.
Although mutational profiling is increasingly used to inform important diagnostic, prognostic, and therapeutic considerations in oncology patients with solid tumors, current clinically used assays are limited in the breadth and depth of detectable alterations. Since 2014, a custom hybridization capture-based next-generation sequencing assay known as IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets) has been used at Memorial Sloan Kettering Cancer Center (MSKCC) (MSK-IMPACT) to comprehensively profile patient tumors at the molecular level, with the capacity to detect somatic alterations in >300 genes at a minimum depth of coverage of 91×.39 To date, >40 000 patient tumors have been profiled by using MSK-IMPACT.40 Given the availability of this platform and the unmet need of improving VTE prediction across the oncologic patient population, we sought to retrospectively analyze solid tumor MSK-IMPACT data to assess molecular determinants of CAT in patients treated at MSKCC.
Methods
Patients and data capture
We included all adult patients who had solid tumor MSK-IMPACT testing between January 2014 and December 2016 as well as available tumor registry and electronic medical record data. All patient samples and data were obtained following MSKCC institutional review board approval. CAT was defined as any instance of pulmonary embolism or lower extremity deep vein thrombosis (DVT), including both proximal and distal DVT events. Symptomatic and asymptomatic VTE episodes were included. Radiology reports from a list of studies susceptible to elicit a diagnosis of pulmonary embolism or lower extremity DVT (supplemental Appendix B, available on the Blood Web site) for the years 2011 to 2016 were searched for key words indicative of VTE (supplemental Appendix C), and all positive reports were reviewed and verified by an observer (S.M.) for the presence of a VTE episode. The pre-IMPACT period from 2011 to 2013 was included to ensure that VTE episodes occurring in the years before cohort entry were identified. The R statistical platform was used along with package TM for the text mining portion of the work. Parallel to this search, pharmacy records were screened for mentions of therapeutic doses of an anticoagulant (supplemental Appendix D), and positive findings were reviewed by an observer (S.M.) for patients not already noted as a case in the radiology text search. All cases discovered in the radiology and anticoagulation search were reviewed and confirmed by a second observer (A.D.). Lastly, the corpus of medical notes ranging the years 2013 to 2016 was processed with a customized natural language processing (NLP) pipeline and data entry interface to identify missing lower extremity DVT and pulmonary embolism cases. Only events having occurred up to 1 year before cancer diagnosis were considered to be cancer related; older episodes were recorded separately. Upper extremity DVT events were captured by using the same customized NLP pipeline with each case reviewed by an observer (J.V.M. and S.M.).
Information on tumor type and basic demographic characteristics was obtained from the institutional tumor registry and clinical database system. To facilitate the analysis and interpretation of the results, tumor type was simplified and limited to 15 categories (supplemental Appendix E). Metastatic status during the study period was estimated by merging staging data from the tumor registry and IMPACT records (supplemental Appendix F). Patients not marked as metastatic upon cohort entry had their clinical notes processed with the NLP pipeline to pick up any missed instances of metastatic disease. A manual audit was conducted on 300 randomly selected patients to assess the accuracy of data collection and aggregation.
MSK-IMPACT sequencing
The MSK-IMPACT assay was performed as previously described.39 Briefly, DNA from both tumors and patient-matched blood samples were obtained. Bar-coded libraries were then generated and sequenced. The custom targeted gene panel consisted of 341 genes. Subsequent versions of MSK-IMPACT contain additional numbers of genes (410 genes in version 2; 468 in version 3; supplemental Appendix G); however, these were ultimately excluded from the final analysis to maintain consistency across the cohort. Mean coverage across all tumor samples was 753×, with a minimum depth of coverage of 91×. Raw results were run through a custom pipeline to identify somatic alterations. Germline mutations were filtered out. Final data from all samples were made publicly available online through the cBioPortal for Cancer Genomics.41,42 For the current analysis, mutation data for all individuals were mapped as binary values (mutated vs unmutated). Only putative driver mutations were retained. Data on copy number alterations and gene fusions were also evaluated. In the case of fusions, only the gene exhibiting a potential change in expression was considered mutated. Fusions detected with the Archer panel (Illumina) were excluded, even though listed in cBioPortal, because only a small number of samples had this assay performed.
Statistical analysis
The primary end point of time-to-CAT was defined as the time from accession to the minimum of CAT development, upper extremity DVT, death, or last follow-up in a 1-year period after accession. CAT included lower extremity DVT (proximal or distal) and pulmonary embolism, symptomatic or not. Accession was defined as the date of blood sample receipt for IMPACT testing and the approximate time a patient consented to testing. Cause-specific Cox proportional hazards regression estimated associations between select somatic mutations and the risk of developing a CAT episode. A separate model was built for each mutation; all models were adjusted for cytotoxic chemotherapy as a time-dependent covariate, history of VTE, current anticoagulation use, presence of metastatic disease, and age at accession. In addition, all models were stratified based on tumor type and, because some tumor samples were previously banked, the years from the procedure to accession ([0], (0-0.25], (0.25-1], (1-5], (5,+]). Left truncation was used for the subset of procedures that occurred after accession but before the end of the 1-year period. We only considered the 50 most commonly mutated genes in the cohort, in addition to MET, ALK, and ROS1, which were selected based on prior data suggesting an effect on the risk of thromboembolism in cancer.22-24,33 The Benjamini-Hochberg method was used to adjust P values for false discovery. The predetermined statistical significance cutoff for the purpose of this analysis was 0.10. The R 3.6.1 statistical platform (R Foundation for Statistical Computing) was used for multivariate analysis.
Results
Patient characteristics and incidence of VTE
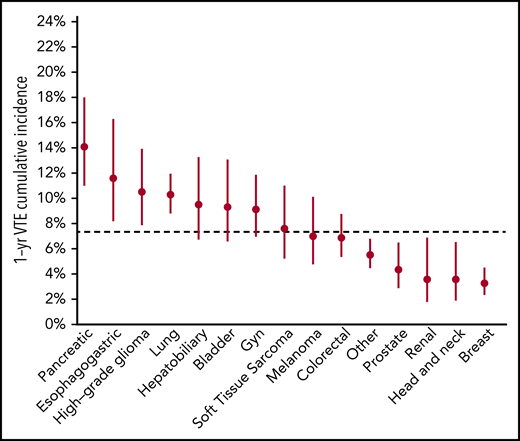
The schema used for VTE assessment in all patients is delineated in supplemental Appendix A and Figure 1. From 2014 to 2016, a total of 14 223 adult patients with solid tumor malignancy had MSK-IMPACT genomic sequencing and met the initial inclusion criteria for this retrospective study. Of these, 1962 were found to have at least one episode of CAT. A total of 2528 patients were excluded from cohort entry due to preexisting CAT (n = 1104), upper extremity DVT (n = 161), or because they had no clinical notes available in the electronic medical record after the start of observation, consisting of the latest of IMPACT tissue sampling or blood sample accession. A total of 11 695 individuals were included in the final analysis, with 693 episodes of CAT recorded in the first year of observation. The highest event rates were in the subgroup of patients with pancreatic cancer (Figure 2). Characteristics of the patient cohort are presented in Table 1. The prevalence of individual cancer types included in the entire IMPACT cohort was generally reflective of the prevalence of cancer types within the larger oncologic patient population; however, breast cancer and prostate cancer, two of the most common cancer types, were underrepresented, likely owing to fewer patients with these tumor types undergoing extended genomic testing during the observed time period. A total of 163 patients (1.4%) had a VTE episode documented >1 year before cancer diagnosis. Importantly, most patients (72%; n = 8383) had metastatic disease at time of MSK-IMPACT testing.
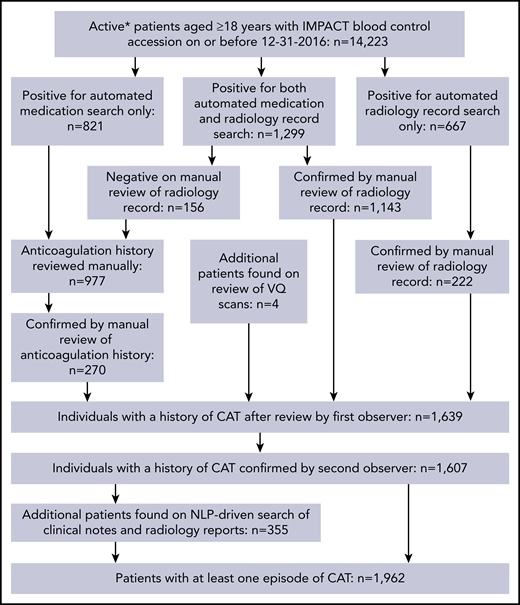
Flow of CAT event assessment. *Patients who were followed up actively at the medical center for the years 2014 to 2016. VQ, ventilation–perfusion.
Flow of CAT event assessment. *Patients who were followed up actively at the medical center for the years 2014 to 2016. VQ, ventilation–perfusion.
Patient characteristics
| Characteristic | Overall | VTE event |
|---|---|---|
| Cytotoxic chemotherapy* | ||
| No | 6759 (58%) | 402 (58%) |
| Yes | 4936 (42%) | 291 (42%) |
| Age at accession, median (IQR), y | 61 (52-70) | 63 (54-71) |
| Anticoagulant subclass | ||
| Dabigatran | 35 (0%) | 3 (0%) |
| Heparinoid | 135 (1%) | 11 (2%) |
| None | 10 925 (96%) | 645 (95%) |
| Vitamin K antagonist | 143 (1%) | 10 (1%) |
| Xa-direct oral anticoagulant | 178 (2%) | 7 (1%) |
| Metastatic disease | ||
| No | 3312 (28%) | 121 (17%) |
| Yes | 8383 (72%) | 572 (83%) |
| Tumor type | ||
| Bladder | 400 (3%) | 32 (5%) |
| Breast | 1690 (14%) | 40 (6%) |
| Colorectal | 1084 (9%) | 62 (9%) |
| Esophagogastric | 286 (2%) | 30 (4%) |
| Gynecologic | 696 (6%) | 51 (7%) |
| Head and neck | 327 (3%) | 10 (1%) |
| Hepatobiliary | 383 (3%) | 32 (5%) |
| High-grade glioma | 487 (4%) | 44 (6%) |
| Lung | 1978 (17%) | 161 (23%) |
| Melanoma | 470 (4%) | 27 (4%) |
| Other | 1894 (16%) | 87 (13%) |
| Pancreatic adenocarcinoma | 487 (4%) | 57 (8%) |
| Prostate adenocarcinoma | 761 (7%) | 24 (3%) |
| Renal | 325 (3%) | 9 (1%) |
| Soft tissue sarcoma | 427 (4%) | 27 (4%) |
| VTE before cancer diagnosis | ||
| No | 11 530 (99%) | 673 (97%) |
| Yes | 165 (1%) | 20 (3%) |
| Characteristic | Overall | VTE event |
|---|---|---|
| Cytotoxic chemotherapy* | ||
| No | 6759 (58%) | 402 (58%) |
| Yes | 4936 (42%) | 291 (42%) |
| Age at accession, median (IQR), y | 61 (52-70) | 63 (54-71) |
| Anticoagulant subclass | ||
| Dabigatran | 35 (0%) | 3 (0%) |
| Heparinoid | 135 (1%) | 11 (2%) |
| None | 10 925 (96%) | 645 (95%) |
| Vitamin K antagonist | 143 (1%) | 10 (1%) |
| Xa-direct oral anticoagulant | 178 (2%) | 7 (1%) |
| Metastatic disease | ||
| No | 3312 (28%) | 121 (17%) |
| Yes | 8383 (72%) | 572 (83%) |
| Tumor type | ||
| Bladder | 400 (3%) | 32 (5%) |
| Breast | 1690 (14%) | 40 (6%) |
| Colorectal | 1084 (9%) | 62 (9%) |
| Esophagogastric | 286 (2%) | 30 (4%) |
| Gynecologic | 696 (6%) | 51 (7%) |
| Head and neck | 327 (3%) | 10 (1%) |
| Hepatobiliary | 383 (3%) | 32 (5%) |
| High-grade glioma | 487 (4%) | 44 (6%) |
| Lung | 1978 (17%) | 161 (23%) |
| Melanoma | 470 (4%) | 27 (4%) |
| Other | 1894 (16%) | 87 (13%) |
| Pancreatic adenocarcinoma | 487 (4%) | 57 (8%) |
| Prostate adenocarcinoma | 761 (7%) | 24 (3%) |
| Renal | 325 (3%) | 9 (1%) |
| Soft tissue sarcoma | 427 (4%) | 27 (4%) |
| VTE before cancer diagnosis | ||
| No | 11 530 (99%) | 673 (97%) |
| Yes | 165 (1%) | 20 (3%) |
IQR, interquartile range.
Cytotoxic chemotherapy anytime from day −30 to day 365 from accession date.
Clinical risk factors were assessed for VTE risk. Consistent with previous reports, cytotoxic chemotherapy (hazard ratio [HR], 1.63; 95% confidence interval [CI], 1.32-1.93; P < .001), prior VTE episode (HR, 2.20; 95% CI, 1.40-3.47; P = .001), and metastatic disease (HR, 2.60; 95% CI, 2.03-3.33; P < .001) were all strongly associated with CAT in multivariate analysis. Similarly, older age, an elevated white blood cell count, decreased hemoglobin, an increased platelet count, and higher body mass index values were associated with a higher risk of CAT. Data on those latter parameters were not available for all patients.
Specific somatic alterations in tumor are associated with increased VTE risk
MSK-IMPACT data were then assessed to determine associations between individual genes and VTE risk across solid tumor malignancies. Tumor molecular profiles were analyzed across all cancer types and within subgroups. Mutational frequencies across tumor types revealed rates of somatic alteration similar to those observed in previously published reports.43 TP53 mutations had the highest prevalence, present in 42% of all tumor samples. KRAS and EGFR mutations were also frequent, found in up to 17% and 6% of tumor samples, respectively, and were identified across all cancer types, with a particularly high incidence in colorectal and pancreatic cancers (supplemental Figure 1). IDH1 mutations were found to be enriched in gliomas (24.9% of these patients), consistent with prior reports.44
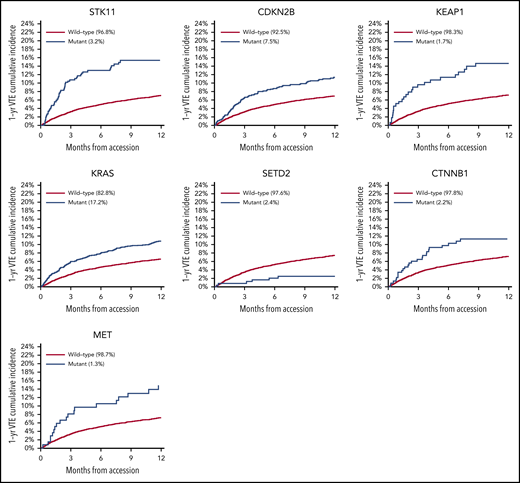
There was no significant association between microsatellite instability and risk of VTE (data not shown). In the multicancer adjusted model, there was no association between tumor mutational burden and VTE risk; however, when cancer types were analyzed individually, significant associations were found in lung and prostate cancer (Figure 3). Each gene was assessed in a separate regression model with the following covariates: age, previous VTE episode, anticoagulation, presence of metastatic disease, and cytotoxic chemotherapy (time dependent). Models were stratified based on tumor type and the time from a prior procedure to blood sample receipt for IMPACT germline control. Notably, mutations in KRAS (HR, 1.34; 95% CI, 1.09-1.64; adjusted P = .08), STK11 (HR, 2.12; 95% CI, 1.55-2.89; adjusted P < .001), KEAP1 (HR, 1.84; 95% CI, 1.21-2.79; adjusted P = .07), CTNNB1 (HR, 1.73; 95% CI, 1.15-2.60; adjusted P = .09), CDKN2B (HR, 1.45; 95% CI, 1.13-1.85; adjusted P = .07), and MET (HR, 1.83; 95% CI, 1.15-2.92; adjusted P = .09) were found to be associated with a significantly increased risk of CAT independent of tumor type (Table 2).
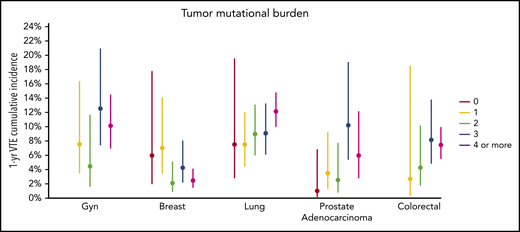
One-year incidence of VTE according to cancer type, stratified by tumor mutational burden. Gyn, gynecologic.
One-year incidence of VTE according to cancer type, stratified by tumor mutational burden. Gyn, gynecologic.
HR of VTE for selected cancer somatic mutations
| Gene* | HR (95% CI) | P | FDR q value |
|---|---|---|---|
| STK11 | 2.12 (1.55-2.89) | .0000023 | <0.001 |
| CDKN2B | 1.45 (1.13-1.85) | .0034719 | 0.072 |
| KEAP1 | 1.84 (1.21-2.79) | .0040963 | 0.072 |
| KRAS | 1.34 (1.09-1.64) | .0058245 | 0.077 |
| SETD2 | 0.35 (0.16-0.79) | .0116198 | 0.088 |
| CTNNB1 | 1.73 (1.15-2.60) | .0089796 | 0.088 |
| MET | 1.83 (1.15-2.92) | .0111671 | 0.088 |
| CDK4 | 1.56 (1.06-2.29) | .025 | 0.148 |
| IDH1 | 0.45 (0.22-0.90) | .024 | 0.148 |
| CDKN2A | 1.26 (1.02-1.55) | .032 | 0.171 |
| SMAD4 | 1.30 (0.96-1.76) | .086 | 0.379 |
| MYC | 1.37 (0.96-1.94) | .080 | 0.379 |
| TP53 | 1.15 (0.98-1.35) | .094 | 0.382 |
| FOXA1 | 0.62 (0.33-1.16) | .133 | 0.505 |
| APC | 0.74 (0.49-1.12) | .159 | 0.561 |
| SMARCA4 | 1.34 (0.87-2.05) | .189 | 0.625 |
| Gene* | HR (95% CI) | P | FDR q value |
|---|---|---|---|
| STK11 | 2.12 (1.55-2.89) | .0000023 | <0.001 |
| CDKN2B | 1.45 (1.13-1.85) | .0034719 | 0.072 |
| KEAP1 | 1.84 (1.21-2.79) | .0040963 | 0.072 |
| KRAS | 1.34 (1.09-1.64) | .0058245 | 0.077 |
| SETD2 | 0.35 (0.16-0.79) | .0116198 | 0.088 |
| CTNNB1 | 1.73 (1.15-2.60) | .0089796 | 0.088 |
| MET | 1.83 (1.15-2.92) | .0111671 | 0.088 |
| CDK4 | 1.56 (1.06-2.29) | .025 | 0.148 |
| IDH1 | 0.45 (0.22-0.90) | .024 | 0.148 |
| CDKN2A | 1.26 (1.02-1.55) | .032 | 0.171 |
| SMAD4 | 1.30 (0.96-1.76) | .086 | 0.379 |
| MYC | 1.37 (0.96-1.94) | .080 | 0.379 |
| TP53 | 1.15 (0.98-1.35) | .094 | 0.382 |
| FOXA1 | 0.62 (0.33-1.16) | .133 | 0.505 |
| APC | 0.74 (0.49-1.12) | .159 | 0.561 |
| SMARCA4 | 1.34 (0.87-2.05) | .189 | 0.625 |
Each gene evaluated in a separate Cox proportional hazards model adjusting for chemotherapy (time-dependent covariate), a history of previous VTE, anticoagulation, presence of metastatic disease, and age at accession. The model is stratified based on tumor type and the time from a prior procedure time to accession.
Mutations in SETD2 were associated with a decreased risk of CAT (HR, 0.35; 95% CI, 0.16-0.79; adjusted P = .09). The unadjusted cumulative incidences of VTE for individuals with vs without a somatic mutation in these 7 genes are shown in Figure 4. The HRs of VTE for a mutation in each gene broken down by tumor type are shown in Figure 5 (also see supplemental Figure 2 for cumulative incidences). The effect of mutations in STK11, CDKN2B, KRAS, and SETD2 was largely consistent across tumor types, whereas the effect of CTNNB1 mutations varied between different histologies. CDK4 and CDKN2A mutations exhibited a trend toward an increased risk of CAT, with no statistically significant effect found after false discovery rate (FDR) adjustment. A decreased risk of CAT was noted with IDH1 mutations, but this effect was not significant after adjustment. Subset analysis was performed for IDH1, stratifying patients according to cancer type. Other than brain gliomas, the only group with a mutational frequency high enough for the analysis consisted of hepatobiliary tumors. Interestingly, the presence of an IDH1 mutation was associated with a lower risk of VTE only for gliomas (HR, 0.26; 95% CI, 0.08-0.79; P = .02), with no significant effect noted in patients with hepatobiliary tumors (HR, 0.81; 95% CI, 0.28-2.37; P = .71). Figure 6 displays 1-year cumulative incidence of VTE in those subgroups. A sensitivity analysis was also performed, excluding patients on anticoagulation. We found similar effects, observing the same direction and magnitude of effect for the 7 genes identified previously (STK11, KEAP1, KRAS, CTNNB1, SETD2, CDKN2B, and MET), even though the resulting q values were above the threshold of 0.1 for SETD2, CDKN2B, and MET.
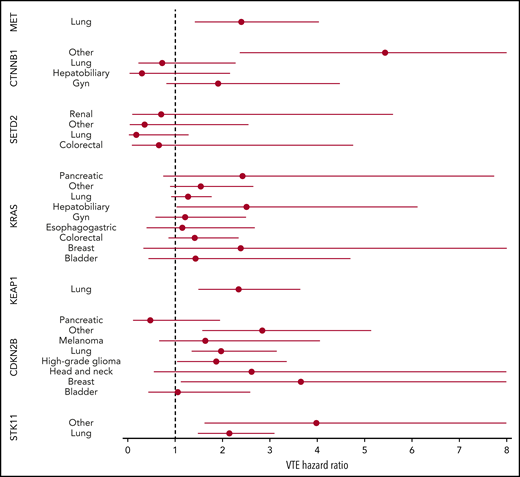
HR of VTE for mutated vs unmutated genes, stratified according to tumor type. Results are shown for groups with at least 25 cases of mutations.
HR of VTE for mutated vs unmutated genes, stratified according to tumor type. Results are shown for groups with at least 25 cases of mutations.
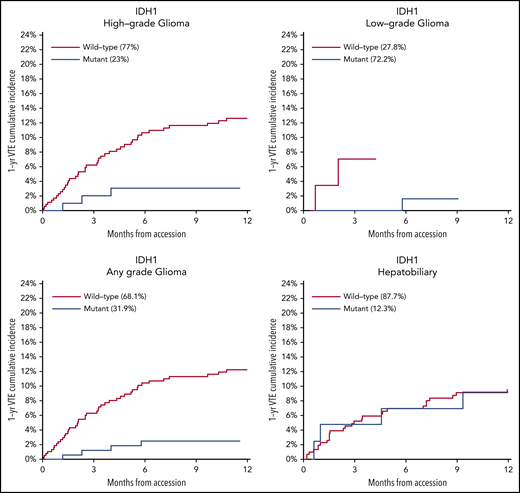
One-year cumulative incidence of CAT according to IDH1 mutation status for gliomas and hepatobiliary tumors. Gyn, gynecologic.
One-year cumulative incidence of CAT according to IDH1 mutation status for gliomas and hepatobiliary tumors. Gyn, gynecologic.
Given the observed effect of STK11 mutations on the risk of CAT, a dedicated analysis was conducted on data from The Cancer Genome Atlas (TCGA) to evaluate the effect of STK11 mutations on RNA expression for tissue factor (F3) and granulocyte colony-stimulating factor (G-CSF) (CSF3). Tissue factor is widely believed to be an important effector of CAT and has been shown to be upregulated by several oncogenes.34 G-CSF has been shown to be a likely mediator of the neutrophilia observed in lung cancer, also potentially being responsible for the formation of neutrophil extracellular traps and thromboembolism.45,46 A data set including 503 patients from TCGA and with lung adenocarcinoma was selected in cBioPortal, given the expected high prevalence of STK11 mutations in this group.41,42,47 RNA expression values were batch normalized (RNA Seq V2 RSEM) and a z score threshold of ±2.0 was used. The messenger RNA expression z scores relative to all samples were 0.58 vs −0.09 for F3 (STK11 mutated vs unmutated; P = 1.334E-07) and 0.39 vs −0.15 for CSF3 (STK11 mutated vs unmutated; P = 9.175E-05). See supplemental Figure 3 for plots generated by cBioPortal. To assess the potential impact of increased transcription of G-CSF, a linear model predicting absolute neutrophil count based on the presence of an STK11 mutation and adjusting for cancer type was fit by using the MSK-IMPACT cohort. The presence of a STK11 mutation was associated with an average increase in 1223 cells/μL for the absolute neutrophil count (P < .001).
Clonal hematopoiesis in solid tumor patients does not increase the risk of VTE
We next assessed MSK-IMPACT data for the presence of CH and association with CAT. Of the 11 695 patients in the final cohort, 30% were found to have CH with a mutation in a known CH gene. There was no significant association between any CH mutations, including JAK2 V617F, and differential risk of CAT in the observed cohort (supplemental Figure 4).
Discussion
This work is the first broad search of a DNA tumor registry of its kind aimed to elucidate cancer-specific genomic determinants of CAT. Identifying mutations that might modify a patient’s risk for CAT may not only improve prognostication but also potentially uncover new pathophysiological mechanisms of how aberrant signaling might lead to VTE development. The results of this study identified multiple individual tumor mutations influencing VTE risk in patients with a solid malignancy, including KRAS, for which previous data exist.20,21,48 Preclinical data show that aberrant signaling through activating mutations of MET might confer a greater risk of CAT,33,49 and this study suggests this finding applies to human neoplasms. In addition, IDH1 mutations in gliomas have been shown to be associated with a lower risk of VTE,31,32 a finding supported by our analysis even though the effect did not reach statistical significance when looking at all tumor types and after adjustment for multiple comparisons. The latter seems to be due to a differential action of IDH1 mutated status across cancer types. In our cohort, there was no evidence of a protective effect from IDH1 mutations against VTE outside of gliomas. This finding is consistent with other data suggesting that the improved vital prognosis associated with IDH mutations (IDH1 and IDH2) in gliomas does not extend to other tumors and that the effects of IDH mutations on modulating VTE risk are mediated or influenced by as-yet unknown cancer type–specific factors, including degree of aberrant tumor hypermethylation.50 If true, future assessments of the effect of IDH1 mutations on the risk of CAT should be conducted within predefined tumor type categories to obtain valid measurements. Parallel to this, the genetic basis of CAT likely varies across tumor types, as mutational patterns are very different between groups, and existing data suggest that expression of circulating mediators responsible for the thrombophilia of cancer vary according to neoplasm as well.51 Also, there is growing evidence that coagulation activation stimulates local cancer progression and metastasis, at least in part due to an effect on tumoral epigenome.19,52 This latter mechanism should be explored in future studies.
Conversely, the increased risk of CAT reported with mutated STK11, CDKN2B, CTNNB1, and KEAP1 genes has not been previously reported. However, the effect of KEAP1 was mitigated in multi-gene regression models (data not shown), and mutations in this gene are strongly correlated with STK11 defects,53 suggesting KEAP1 mutations might not be an independent predictor of CAT risk. The mechanisms by which STK11 mutations are associated with an increased risk of VTE are unclear and might include increased neutrophil extracellular trap formation secondary to G-CSF production by the tumor. The possible explanations behind the decreased risk of CAT seen with SETD2 mutations also remain to be determined. The effect of mutations in STK11, CDKN2B, KRAS, and SETD2 was largely consistent across tumor types, and in multivariate analyses, all HRs were adjusted for cancer type. This suggests that the gene–thrombosis association for these genes is not simply an effect of the tumors with which they are associated but rather a genuine downstream biological effect of altered gene-specific signaling.
Notably, previous studies found conflicting results with regard to EGFR mutational status in lung cancer and VTE risk, with either a protective effect, no effect, or an increased risk.20,25-28 In this large retrospective cohort including most solid tumor types, we failed to establish a significant association between CAT risk and the presence of an EGFR mutation. One possible explanation would be that in the last few years, patients with EGFR-mutated lung cancer have received EGFR-targeted therapies. In this regard, anti-EGFR monoclonal antibodies have been associated with an increased risk of arterial and VTE, whereas EGFR-targeted tyrosine kinase inhibitors have not.54,55 As such, differences in exposure to EGFR-directed therapies in different cohorts of patients with lung cancer could potentially result in discordant estimates of thromboembolism risk.
Available peripheral blood sequencing data for this large cancer cohort also allowed for the detection of CH and assessment of its effect on CAT risk. No significant association was found between the presence of any CH mutation and elevated VTE risk across all tumor types, including JAK2 V617F, despite a previously published report of this association in non-cancer patient populations.38 Germline mutations were also excluded from this analysis.
Any retrospective cohort study such as this is prone to several potential pitfalls in terms of data collection and analysis. Ascertainment of clinical events on a cohort of >10 000 individuals is particularly challenging. In the current case, only a fraction of the available clinical notes were reviewed by a human observer, and thus it is possible that a small number of CAT events were missed. In addition, another important aspect in determining the validity of results for a clinical genomic study is the method of classifying genetic data with respect to mutational status. We decided to use a simple approach, labeling individual cohort members as being mutated or not for any given gene. One might devise a more sophisticated method and subclassify participants based on the specific mutations encountered for the most commonly mutated genes such as KRAS.
It is also worth noting that mutational status influencing treatment choices could potentially result in spurious associations between genotype and VTE risk. In such a retrospective study, further grouping of treatment types into specific cytotoxic chemotherapy subtypes or considering other therapies would considerably complicate the model. Future work elucidating the biological basis for a change in CAT risk associated with any given mutation will be needed to confirm the associations we reported. Finally, estimation of the FDR is another very important aspect of cancer genomic studies. We chose 0.10 as a cutoff for significance due to the exploratory nature of the study; if the more stringent level of 0.05 was applied, only STK11 would be considered significant. It is also very possible that some genes with an FDR >0.10 in our models might be significant predictors of CAT. In this regard, any analysis measuring multiple associations requires special care to interpret results of statistical significance tests. How these mutations might contribute to increased thrombosis development is not known and will require further validation along with functional studies.
In summary, improved risk stratification methods for VTE risk are needed for the diagnosis and prevention of CAT. Enhanced genetic tools allow for the detection of molecular events that might contribute to CAT risk. Retrospective research such as the work presented here can help elucidate the pathophysiology of CAT and aid in the development of risk stratification tools; however, the high dimensionality of the data sets used is challenging. The analysis we performed on a large cohort of patients with solid tumors suggests associations between cancer-specific mutations and CAT risk, justifying further validation of key results along with dedicated functional studies to better elucidate how tumor-specific alterations contribute to thrombotic disease in this high-risk population.
Original data may be requested by contacting the corresponding author (Simon Mantha; e-mail: manthas@mskcc.org).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
Acknowledgments
The results published here are based, in part on data generated by the TCGA Research Network (https://www.cancer.gov/tcga).
Memorial Sloan Kettering research support was provided by the National Institutes of Health (NIH), National Cancer Institute (NCI) Cancer Center Support Grant (P30 CA008748). A.D. receives support from the American Association of Cancer Research (17-40-11-DUNB). A.A.K. acknowledges additional research support from the Sondra and Stephen Hardis Chair in Oncology Research and the NIH, National Heart, Lung, and Blood Institute (U01HL143402). Y.P. receives funding from the NIH, NCI (K12 CA184746 Paul Calabresi Award), the Parker Institute for Cancer Immunotherapy, and the Society for Immunotherapy of Cancer Sparkathon TimIOs project. N.M.I. receives research support from the NIH, NCI (R01CA235711), the Breast Cancer Research Foundation, and the American Cancer Society. J.G. was supported by the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Authorship
Contribution: S.M. planned the study and contributed to data collection and analysis and drafting of the manuscript; A.D. drafted the manuscript and helped oversee the analysis; S.M.D. performed the statistical analysis; K.L.B. contributed to the analysis and drafting of the manuscript; J.V.M., M.F., K.B.C., and Y.P. helped with data collection; F.S.-V. and J.G. contributed to the analysis; K.J., J.W., and D.K. helped with data aggregation; N.M.I. contributed to planning of the study; and R.L.L., S.K., A.Z., W.P., A.A.K., and G.A.S. contributed to drafting of the manuscript.
Conflict-of-interest disclosure: S.K. serves as a consultant to Inari Medical. R.L.L. is on the supervisory board of Qiagen; is a scientific advisor to Loxo, Imago, C4 Therapeutics, and Isoplexis, each of which includes equity interest; receives research support from and consulted for Celgene and Roche; received research support from Prelude Therapeutics; has consulted for Lilly, Janssen, Incyte, Novartis, and Gilead; and has received honoraria from Lilly and Amgen for invited lectures. W.P. receives funding from Merck, Astellas, and Gossamer Bio; and is a consultant to Ipsen. A.A.K. received fees from Janssen, Bayer, Sanofi, Parexel, Halozyme, Pfizer, Seattle Genetics, Pharmacyclics, PharmaCyte, AngioDynamics, Leo Pharma, TriSalus, and Medscape; and grant support from Merck, Array, Bristol Myers Squibb, and Leap. S.M. is principal owner of Daboia Consulting LLC; and received speaking fees from MJH Associates and Physicians’ Education Resource. J.V.M. received fees from Sobi/Dova Pharmaceuticals. N.M.I. received consulting fees from Novartis and Seattle Genetics, along with grant support from Novartis. The remaining authors declare no competing financial interests.
Correspondence: Simon Mantha, Memorial Sloan Kettering Cancer Center, Koch Center, 545 East 73rd St, New York, NY 10021; e-mail: manthas@mskcc.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal