

Is abnormal clotting in a cancer patient an unspecific aftermath of tissue and vascular damage, inflammation, or treatment, or is it an intrinsic part of the disease “program” embedded in its oncogenic driver genes? Can there be tumor-specific causes, predictors, and possibly treatments of cancer-associated thrombosis (CAT)? These seemingly simple questions baffled investigators for decades, while CAT continued to exact a toll of human suffering and loss of life, as one of the leading causes of cancer-related mortality.1,2 In this issue of Blood, embark on a bold attempt to seek some of the possible answers.3
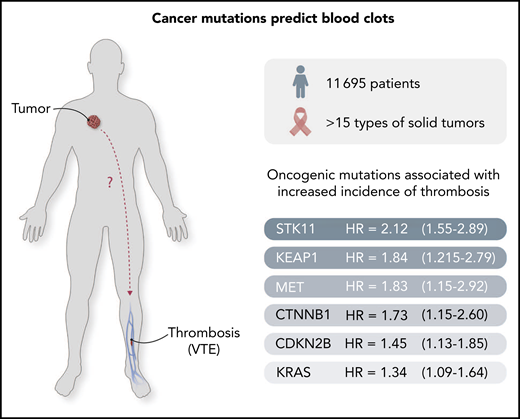
Cancer-causing mutations predict clotting risk. A set of oncogenic mutations has been identified3 to predict the risk of thrombosis in cancer patients. The outstanding challenge is to uncover the mechanisms whereby these mutant cancer genes project their influence systemically, as shown by the "?" in the figure.
Cancer-causing mutations predict clotting risk. A set of oncogenic mutations has been identified3 to predict the risk of thrombosis in cancer patients. The outstanding challenge is to uncover the mechanisms whereby these mutant cancer genes project their influence systemically, as shown by the "?" in the figure.
The authors document a striking link between cancer-specific (oncogenic) mutations in cancer cells and the incidence of CAT in a large cohort of 11 695 cancer patients. Remarkably, they report that regardless of the tumor type, mutations in certain genes, such as STK11, KRAS, CTNNB1, KEAP1, CDKN2B, and MET, predict the significantly elevated incidence of CAT for up to 1 year before diagnosis, whereas other genes are not predictive, or even seem to exhibit a negative (protective) association (SETD2, IDH1).3 This is the first such large-scale effort to explore the link between cancer genomics and thrombosis, and the results are thought provoking.
The notion that certain oncogenic mutations may deregulate hemostatic genes (coagulome) in cancer cells has been entertained for some time,4,5 but the related studies were either experimental4,5 or focused on limited cohorts of patients with specific cancers.6,7 Some of these analyses also emphasized specific oncogenes or coagulant effectors, for instance, tissue factor,4 or applied correlative gene-finding algorithms.8 Although stimulating curiosity, these efforts could not have successfully challenged the prevailing view that solutions to the daunting problem of cancer-related venous thromboembolism (VTE) and its morbid, often life-threatening manifestations, such as deep vein thrombosis or pulmonary embolism, reside principally within the hemostatic system.
This blood-centric perspective is put to the test by Dunbar and colleagues through a deliberate application of a tumor-centric approach, in which cancer-driving mutations are tested for their link to the VTE. The authors deployed an awesome power of a large cancer patient registry compiled at Memorial Sloan Kettering Cancer Center, where the genomes of thousands of cancer specimens are routinely sequenced using a panel of ≥341 selected genes. This database was carefully filtered, resulting in retention of 11 695 cases, including >15 different solid tumor types, mostly (72%) in a metastatic stage of progression. Among these patients, there were 693 episodes of CAT, which the authors proceeded to query for associations with standard clinical variables (diagnosis, stage, therapy) and, most importantly, for the linkage with oncogenic mutations. Remarkably, several genes were found to be significantly associated with the VTE risk, regardless of tumor type. In some cases (STK11/LKB1), these associations withstood a stringent statistical scrutiny, whereas in other cases, they became weaker or nonexistent outside of a specific type of malignancy or context. For example, IDH1 mutations were (as shown earlier7 ) protective of thrombosis in glioma, but not in hepatocellular carcinoma. Notably, mutations with a widespread effect on the epigenome, such as those affecting SETD2 (histone methyltransferase), were found to be negatively associated with VTE in this cohort.
The authors also interrogated genetic aberrations occurring in the “background” of active cancers and manifested as clonal hematopoiesis (CH), especially in older patients. However, CH did not seem to impact the VTE risk. They also explored correlations between stronger genetic “hits,” such as mutant STK11 searching gene expression databases for putative biological mediators of CAT. Interestingly, upregulation of 2 of such candidates: tissue factor (TF) and granulocyte colony-stimulating factor (G-CSF), was found in the subset of lung cancer patients with tumors harboring mutant STK11, along with increased leukocyte counts already linked with thrombosis.9
This is an important study for at least 2 reasons. First, it exponentially enlarges the suggested4 link between the cancer genome and disease-related hemostatic perturbations. The authors duly acknowledge the contribution of clinical variables to the incidence of CAT, but point to the component of tumor-specific oncogenic pathways superimposed upon the disease etiology. Second, the study paves the way for studies of molecular interplay between cancer and blood compartments involved in triggering thrombosis, with hopes to develop more precise predictive algorithms, mutation-informed diagnostics, and interventions in CAT/VTE. Because thrombosis is not only morbid in its own right, but also may influence cancer biology,4 the genetic link in this chain of events, especially established on a large clinical scale, is of considerable interest.
Although the work of Dunbar touches on some pivotal questions, it also leaves several of them unanswered, perhaps as any solid scientific inquiry should. For example, the central puzzle in the pathogenesis of VTE is how a tumor with defined organ location, even if metastatic, can trigger thrombosis in a distant vascular bed (see figure) and in a manner related to its genetic makeup? One answer that has been contemplated for some time is that cancer cells may project their coagulant effect systemically through the release into the circulation of soluble mediators (eg, G-CSF) with indirect effects, or by shedding coagulant extracellular vesicles (EVs) harboring TF or podoplanin.4,5,9,10 Interestingly, EVs (microparticles) are, indeed, impacted by oncogenic transformation,4,5 and it would be truly fascinating to explore the related effects of mutant STK11. Another pressing question relates to the interplay between different mutations within specific tumor subtypes. For example, KRAS and TP53 coregulate TF expression and its EV-mediated release in colon cancer cells,4 and the extent of such multigenic regulation in specific disease settings could make the prediction of VTE/CAT even more precise. Finally, some of the oncogenes implicated in this study as correlates (if not triggers) of CAT are known therapeutic targets (eg, MET). It is of interest whether drugs directed at these transforming proteins might alter the course of CAT, and conversely, whether targeting “protective” mutations, such as SETD2 or IDH1 (in glioma), may come at a cost of thrombosis. These and other questions are presently unanswered, but they might not have been asked, if not for the present study, which will undoubtedly shape the way we think about cancer-related thrombosis.
Conflict-of-interest disclosure: The author declares no competing financial interests.
