

Key Points
Targeting 1% to 3% and 8% to 12% FVIII troughs was efficacious, with fewer bleeds in the latter arm and acceptable safety across arms.
Rurioctocog alfa pegol consumption varied widely in each arm with overlapping ranges, emphasizing the need for personalized treatment.
Visual Abstract

Abstract
Rurioctocog alfa pegol prophylaxis targeting factor VIII (FVIII) troughs ≥1% has shown to be efficacious with an acceptable safety profile in people with hemophilia A (PwHA). The PROPEL trial compared safety and efficacy of 2 target FVIII troughs in PwHA aged 12 to 65 years, with severe disease, annualized bleeding rate ≥2, and previous FVIII treatment. PwHA were randomized to 12 months’ pharmacokinetic (PK)-guided rurioctocog alfa pegol prophylaxis targeting FVIII troughs of 1% to 3% (reference arm) or 8% to 12% (elevated arm); first 6 months was treatment-adjustment period. The primary endpoint was absence of bleeds during the second 6 months, analyzed using multiple imputations (full analysis set [FAS]). In the 1% to 3% and 8% to 12% arms, respectively, point estimates (95% confidence interval) of proportions of PwHA with zero total bleeds were 42% (29% to 55%) and 62% (49% to 75%) in FAS (N = 115; P = .055) and 40% (27% to 55%) and 67% (52% to 81%) in per-protocol analysis set (N = 95; P = .015). Dosing frequency and consumption varied in each arm. Adverse events (AEs) occurred in 70/115 (60.9%) PwHA; serious AEs in 7/115 (6%) PwHA, including 1 treatment-related in 8% to 12% arm (transient anti–FVIII inhibitor). There were no deaths, serious thrombotic events, or AE-related discontinuations. PK-guided prophylaxis was achievable and efficacious in both arms. No new safety signals were observed in the 8% to 12% arm. These results demonstrate elevated FVIII troughs can increase the proportion of PwHA with zero bleeds and emphasize the importance of personalized treatment. This trial was registered at www.clinicaltrials.gov as #NCT02585960.
Introduction
Standard factor VIII (FVIII) prophylaxis for treatment of hemophilia A is based on patient weight, severity of FVIII deficiency, and bleeding patterns (eg, location/extent of breakthrough bleeding, joint status), with treatment dose and frequency adjusted according to a patient’s clinical response.1 The FVIII concentrate dose required to achieve a desired plasma FVIII level varies among individuals owing to differences in the pharmacokinetic (PK) profile of FVIII coagulant activity.2-4 An individualized approach to prophylaxis, which can optimize treatment and improve outcomes, takes into consideration a patient’s PK profile, phenotypic bleeding pattern, and other factors, such as perceived risk of injury-related bleeds.1,5,6 Evidence suggests that targeting a 1% FVIII trough level may not prevent bleeding episodes in all people with hemophilia A (PwHA).2 In addition, annual number of joint bleeds has been shown to approach zero in PwHA with baseline FVIII levels ≥10%,7 and bleeding rates in those receiving prophylaxis fall as FVIII levels increase.8 A PK-guided regimen where FVIII troughs were sustained above the ∼10% threshold was therefore investigated.
This phase 3, randomized study investigated the efficacy and safety of PK-guided prophylaxis with rurioctocog alfa pegol (TAK-660, SHP660, BAX 855; ADYNOVATE [US]/ADYNOVI[Europe]; Baxalta US Inc, a Takeda company, Lexington, MA)9-13 targeting FVIII trough levels of 1% to 3% and 8% to 12%. The aim was to determine the impact of targeting elevated FVIII troughs and related increased exposure to total FVIII over time on clinical outcomes in PwHA.
Methods
Trial summary
PROPEL, a phase 3, prospective, randomized, open-label, multicenter clinical study compared safety and efficacy of rurioctocog alfa pegol following PK-guided prophylaxis targeting 2 FVIII troughs in patients with severe hemophilia A (clinicaltrials.gov #NCT02585960; EudraCT 2014-005477-37). It was conducted at 62 study sites in 19 countries from November 2015 to August 2018. The protocol was approved by the independent review board at each participating site, and the study was conducted in compliance with the Declaration of Helsinki and Good Clinical Practice Guidelines of the International Conference on Harmonization.
Patients
The study enrolled PwHA 12 to 65 years of age with severe hemophilia A (FVIII level <1%), and an annualized bleeding rate (ABR) ≥2 during the 12 months before study entry. Enrolled PwHA had either completed a previous rurioctocog alfa pegol study11-14 or were naive to rurioctocog alfa pegol and had received prophylaxis or on-demand treatment (for breakthrough bleeds) with plasma-derived or recombinant FVIII for ≥150 documented exposure days. Exclusion criteria included confirmed FVIII inhibitory antibodies ≥0.6 Bethesda units (BU)15 before or at screening, and any inherited or acquired hemostatic defect other than hemophilia A. All PwHA provided written informed consent. Full eligibility criteria are presented in the supplemental methods, available on the Blood Web site.
Patient data collected at screening included presence of ≥4 spontaneous bleeds into a single joint within the previous 6-month period. Also, central laboratory assessments included viral serology for HIV-1/2 antibody and hepatitis C virus (HCV) antibody. Patients positive for HCV antibody underwent polymerase chain reaction testing to confirm no active infection was present.
Study design and treatment
Upon confirmation of eligibility, PwHA underwent a 72- to 96-hour washout period followed by initial PK parameter evaluation. Blood samples were taken within 30 minutes before a single IV infusion of 60 ± 5 IU/kg rurioctocog alfa pegol and at 7 time points up to 96 ± 4 hours postinfusion.
PwHA were subsequently randomly assigned (1:1) to 12 months of PK-guided prophylaxis with rurioctocog alfa pegol targeting FVIII troughs of either 1% to 3% (reference arm) or ∼10% (8% to 12%; elevated arm; Figure 1). Randomization was stratified according to prestudy treatment regimen and ABR (prophylaxis with ABR <5 vs prophylaxis with ABR ≥5 vs on-demand) and was independent of the patient’s PK profile.
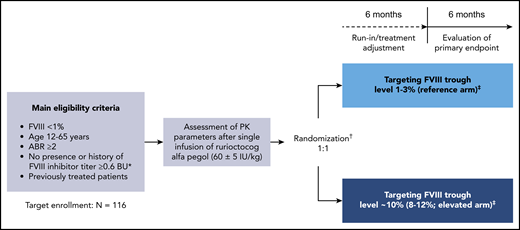
PROPEL study design. Design of this phase 3, prospective, randomized, open-label, multicenter clinical study (#NCT02585960); initiated November 2015, completed August 2018. *In the Nijmegen modification of the Bethesda assay. †Randomization occurred after the PK assessment and was stratified according to patients’ prestudy treatment regimen and ABR (prophylaxis with ABR <5 vs prophylaxis with ABR ≥5 vs on-demand) independent of their individual characteristics (ie, PK profile, activity level, or bleeding activity). ‡Prophylactic dose and dosing frequency with rurioctocog alfa pegol were based on the patient’s individual PK assessment and could be adjusted to maintain the target FVIII trough level on the basis of FVIII assessments at each study visit during the first 6-month study period.
Each patient was provided with a diary in electronic/paper format to record details of their infusions, bleeds and response to treatment, physical activity, unexpected events, and patient-reported outcomes. Definition of a bleeding event is provided in the supplemental methods. Adherence to the individualized prophylaxis regimen over the 12-month study period (adherence to schedule for ≥75% of prophylactic infusions and adherence to required dose [±10%] for ≥75% of prophylactic infusions) was determined based on patient-reported records from study diaries. Unexpected events recorded in the diary were reported as adverse events (AEs) at the investigator’s discretion.
Outcome measures
The primary efficacy outcome measure was presence or absence of any bleeds in the second 6-month study period. These results are reported as proportion of PwHA with zero total bleeds, which represents all spontaneous and injury-related bleeds that were treated or not treated (ie, all bleeds).
Secondary efficacy outcome measures included proportions of PwHA who achieved zero spontaneous bleeds and zero spontaneous joint bleeds during the second 6 months. In addition, total ABR, spontaneous ABR, spontaneous joint ABR, joint ABR, ABR in joints with ≥4 spontaneous bleeds in 6 consecutive months, and injury-related ABR were calculated. Total ABR during the 12-month study period was compared with historical ABR during the 12 months before enrollment. Physical activity was assessed based on the change in number of days with >15 minutes of physical activity (mild, moderate, strenuous) participation. Weight-adjusted consumption of rurioctocog alfa pegol and frequency of prophylactic infusions during the second 6 months were evaluated.
Safety was assessed by reporting of AEs during the randomized period (excluding surgery periods) and changes in vital signs and clinical laboratory parameters. AEs were categorized according to the Medical Dictionary for Regulatory Activities (MedDRA, version 21.0). Immunogenicity of rurioctocog alfa pegol was measured pre- and postinfusion and when clinically indicated. Inhibitory antibodies to FVIII were measured at a central laboratory.15 Binding antibodies to FVIII, rurioctocog alfa pegol, polyethylene glycol (PEG), and anti–Chinese hamster ovary protein were measured with enzyme-linked immunosorbent assay in compliance with regulatory guidelines16,17 and following principles described by Whelan et al.18
PK parameters for rurioctocog alfa pegol based on FVIII activity included plasma t1/2 (hours) and IR (IU/dL / IU/kg), and average FVIII activity (IU/dL) over time according to 1-stage clotting assay. Testing was performed at a central laboratory (assays and instruments used are reported in the supplemental methods).
Post hoc analyses included evaluation of ABRs in the per-protocol analysis set (PPAS) during the second 6 months according to preenrollment regimen (on-demand treatment or prophylaxis). Also, clinical course of spontaneous bleeds into the same joint of treated PwHA with ≥6 months of observation was evaluated (presence of ≥4 vs <4 spontaneous bleeds into the same joint; PPAS).
Statistical analysis
For calculation of sample size, rates of bleed-free PwHA in 6 months were assumed to be 40% and 70% for target FVIII troughs of 1% to 3% and 8% to 12%, respectively, based on modeling results from the pivotal rurioctocog alfa pegol trial.11 Based on these assumptions, an estimated 48 PwHA per treatment arm were needed to reject the null hypothesis of no difference between arms against a 2-sided alternative at the 5% level of statistical significance with 80% power. Assuming a dropout rate of ∼10% and noncompliance rate of 10% to 15%, 116 PwHA were required to meet statistical power.
The full analysis set (FAS) comprised all randomized PwHA who received prophylaxis for any time period. The PPAS comprised all PwHA in the FAS who completed the full 364-day study period of prophylaxis and had no significant protocol deviations. The safety analysis set (SAS) comprised all enrolled PwHA with ≥1 rurioctocog alfa pegol infusion. The PK analysis set comprised all PwHA in the SAS with ≥1 quantifiable postdose FVIII activity level without significant protocol deviations or events with potential to affect PK analysis.
For the primary efficacy outcome measure, proportions of PwHA with zero total bleeds during the second 6 months in the 2 treatment arms were compared using a χ2 test with continuity correction at a 2-sided 5% level of significance. Data analyses for the primary efficacy outcome followed the intent-to-treat principle, and missing bleed data were imputed for PwHA without 364 days of observation using a multiple imputation technique.19 ABR for secondary efficacy outcomes did not include imputed bleeds and was calculated as: ABR = (observed number of bleeds)/(period under consideration in years).
For all other outcome measures, descriptive statistics were used to analyze the 2 treatment arms. Point estimates (means, medians) and their standard deviations (SDs) and quartiles were calculated for weight-adjusted consumption of rurioctocog alfa pegol and number of rurioctocog alfa pegol infusions for treatment of a bleed. PK parameters by prophylactic regimen are summarized descriptively. All analyses were performed using SAS (SAS Institute Inc, Cary, NC), version 9.4.
Results
Study population
Overall, 135 PwHA were enrolled, and 120 received a single PK dose of 60 ± 5 IU/kg rurioctocog alfa pegol (Figure 2). Fifteen PwHA did not receive the initial PK dose because of screen failures (n = 7) and discontinuations (n = 8), and 5 PwHA discontinued after the initial PK dose and before the first prophylactic dose; thus, 115 PwHA were randomized to the target 1% to 3% FVIII trough (n = 57) or 8% to 12% FVIII trough (n = 58) and received ≥1 prophylactic dose (FAS). A total of 8 PwHA discontinued from the 8% to 12% arm (5 during the first 6 months and 3 during the second 6 months) vs 1 person who discontinued from the 1% to 3% arm during the second 6 months. The documented reason for the 1 discontinuation in the 1% to 3% arm was withdrawal by patient (reason not provided). In the 8% to 12% arm, documented reasons for discontinuation were withdrawal by physician (n = 1; patient did not record in the eDiary daily), withdrawal by patient (n = 3), withdrawal by sponsor (n = 1), noncompliance to study procedures (n = 2), and use of another FVIII because of temperature excursion of study drug (n = 1). The 3 withdrawals by patients in the 8% to 12% arm were documented as poor IV access and required daily infusion (n = 1), refusal to agree to informed consent (n = 1), and noncompliance to study protocol (n = 1). A total 5/57 PwHA in the 1% to 3% arm and 15/58 PwHA in the 8% to 12% arm of the FAS were excluded from the PPAS. The PPAS comprised 95 PwHA (n = 52, 1% to 3% arm; n = 43, 8% to 12% arm). Reasons for exclusion from the PPAS were failure to complete the full 364-day study period of prophylaxis (n = 5; n = 10) and significant protocol deviations (n = 0; n = 5; Figure 2). In the 8% to 12% arm, significant protocol deviations were patient did not administer rurioctocog alfa pegol for 50 days (n = 1), patient did not follow dose adjustments (n = 1), patients were not compliant with rurioctocog alfa pegol (n = 2), and patient administered <75% of planned total dose (n = 1).
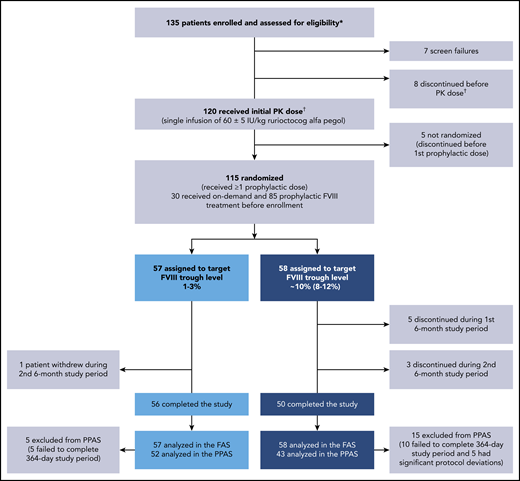
Patient disposition. A total of 135 patients enrolled in the study: 120 patients received the initial PK dose of rurioctocog alfa pegol and 115 patients received at least 1 prophylactic dose of rurioctocog alfa pegol. *Patients either completed a previous rurioctocog alfa pegol study (#NCT01599819,11 #NCT01736475,11 #NCT02210091,13 #NCT02615691, #NCT01913405,12 or #NCT0194559314 ) or were naive to rurioctocog alfa pegol. †One patient in the screen failure category erroneously received the PK infusion and, although not included in the flowchart, is included in the safety analysis and PK analysis sets.
All PwHA were male, and the median (range) age of the overall population was 29.0 (12 to 61) years (FAS and PPAS). At baseline, there was a similar distribution of PwHA who had 1 to 3 joints with ≥4 spontaneous bleeds each within 6 months before screening across both treatment arms. However, there was a higher percentage of PwHA in the 8% to 12% vs 1% to 3% arm who had ≥4 joints with ≥4 spontaneous bleeds each and a higher percentage of PwHA with hemophilic arthropathy. In the FAS, prophylactic FVIII replacement therapy was previously used by 75.4% (43/57) and 72.4% (42/58) of PwHA in the 1% to 3% and 8% to 12% arms, respectively (Table 1); similar results were observed in the PPAS.
Patient demographics and baseline characteristics
| FAS (N = 115) | PPAS (N = 95) | |||
|---|---|---|---|---|
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 58) | FVIII trough level 1% to 3% (n = 52) | FVIII trough level 8% to 12% (n = 43) | |
| Age, y | ||||
| Mean (SD) | 31.1 (13.8) | 31.2 (12.2) | 31.0 (13.6) | 31.6 (12.9) |
| Median (range) | 29.0 (12-61) | 30.5 (13-61) | 28.5 (12-61) | 31.0 (13-61) |
| Sex, n (%) | ||||
| Male | 57 (100) | 58 (100) | 52 (100) | 43 (100) |
| Race, n (%) | ||||
| White | 40 (70.2) | 36 (62.1) | 36 (69.2) | 25 (58.1) |
| Asian | 14 (24.6) | 18 (31.0) | 13 (25.0) | 15 (34.9) |
| Other | 3 (5.3) | 4 (6.9) | 3 (5.8) | 3 (7.0) |
| Previous use of FVIII replacement therapy, n (%) | ||||
| Prophylaxis | 43 (75.4) | 42 (72.4) | 39 (75.0) | 30 (69.8) |
| On-demand | 14 (24.6) | 16 (27.6) | 13 (25.0) | 13 (30.2) |
| Number of joints with ≥4 spontaneous bleeds within 6 mo before screening, n (%) | ||||
| 0 | 17 (29.8) | 14 (24.1) | 16 (30.8) | 10 (23.3) |
| 1 | 15 (26.3) | 17 (29.3) | 14 (26.9) | 15 (34.9) |
| 2 | 15 (26.3) | 13 (22.4) | 12 (23.1) | 7 (16.3) |
| 3 | 5 (8.8) | 4 (6.9) | 5 (9.6) | 2 (4.7) |
| ≥4 | 5 (8.8) | 10 (17.2) | 5 (9.6) | 9 (20.9) |
| Hemophilic arthropathy present at screening, n (%)* | 7 (12.3) | 15 (25.9) | 7 (13.5) | 12 (27.9) |
| HCV antibody present, n (%)† | 30 (52.6) | 26 (44.8) | 27 (51.9) | 21 (48.8) |
| HIV-1/2 antibody present, n (%) | 2 (3.5) | 6 (10.3) | 2 (3.8) | 4 (9.3) |
| ABR (all bleeds) within 12 mo before enrollment‡ | ||||
| Mean (SD) | 13.3 (15.9) | 13.3 (17.4) | 11.8 (14.0) | 14.3 (18.5) |
| Median (Q1 to Q3) | 6.0 (3.0-14.0) | 5.0 (2.0-16.0) | 6.0 (3.0-13.0) | 7.0 (2.0-20.0) |
| FAS (N = 115) | PPAS (N = 95) | |||
|---|---|---|---|---|
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 58) | FVIII trough level 1% to 3% (n = 52) | FVIII trough level 8% to 12% (n = 43) | |
| Age, y | ||||
| Mean (SD) | 31.1 (13.8) | 31.2 (12.2) | 31.0 (13.6) | 31.6 (12.9) |
| Median (range) | 29.0 (12-61) | 30.5 (13-61) | 28.5 (12-61) | 31.0 (13-61) |
| Sex, n (%) | ||||
| Male | 57 (100) | 58 (100) | 52 (100) | 43 (100) |
| Race, n (%) | ||||
| White | 40 (70.2) | 36 (62.1) | 36 (69.2) | 25 (58.1) |
| Asian | 14 (24.6) | 18 (31.0) | 13 (25.0) | 15 (34.9) |
| Other | 3 (5.3) | 4 (6.9) | 3 (5.8) | 3 (7.0) |
| Previous use of FVIII replacement therapy, n (%) | ||||
| Prophylaxis | 43 (75.4) | 42 (72.4) | 39 (75.0) | 30 (69.8) |
| On-demand | 14 (24.6) | 16 (27.6) | 13 (25.0) | 13 (30.2) |
| Number of joints with ≥4 spontaneous bleeds within 6 mo before screening, n (%) | ||||
| 0 | 17 (29.8) | 14 (24.1) | 16 (30.8) | 10 (23.3) |
| 1 | 15 (26.3) | 17 (29.3) | 14 (26.9) | 15 (34.9) |
| 2 | 15 (26.3) | 13 (22.4) | 12 (23.1) | 7 (16.3) |
| 3 | 5 (8.8) | 4 (6.9) | 5 (9.6) | 2 (4.7) |
| ≥4 | 5 (8.8) | 10 (17.2) | 5 (9.6) | 9 (20.9) |
| Hemophilic arthropathy present at screening, n (%)* | 7 (12.3) | 15 (25.9) | 7 (13.5) | 12 (27.9) |
| HCV antibody present, n (%)† | 30 (52.6) | 26 (44.8) | 27 (51.9) | 21 (48.8) |
| HIV-1/2 antibody present, n (%) | 2 (3.5) | 6 (10.3) | 2 (3.8) | 4 (9.3) |
| ABR (all bleeds) within 12 mo before enrollment‡ | ||||
| Mean (SD) | 13.3 (15.9) | 13.3 (17.4) | 11.8 (14.0) | 14.3 (18.5) |
| Median (Q1 to Q3) | 6.0 (3.0-14.0) | 5.0 (2.0-16.0) | 6.0 (3.0-13.0) | 7.0 (2.0-20.0) |
Status based on the patient’s medical history (not a determination made by the investigator on the basis of their clinical evaluation of the patient). No other testing was done in this protocol to establish arthropathy.
HCV antibody-positive patients underwent polymerase chain reaction testing to confirm that no active infection was present.
For newly recruited PwHA, ABRs were assessed on the basis of documented and treated bleeds within 12 mo before enrollment, using patients’ medical records.
Prophylactic dosing intervals used by study completion ranged from every 24 to 96 hours. In the 1% to 3% and 8% to 12% arms, respectively, 0% and 12.1% (7/58) of PwHA administered doses every 24 hours, 19.3% (11/57) and 60.3% (35/58) every 48 hours, 19.3% (11/57) and 12.1% (7/58) every 72 hours, 40.4% (23/57) and 0% every 84 hours, 14.0% (8/57) and 1.7% (1/58) every 96 hours, and 7.0% (4/47) and 0% alternated between 72 and 96 hours (FAS; supplemental Table 1). Of 7 patients in the 8% to 12% arm who received daily infusions, 6 completed the study.
There was no difference in physical activity from baseline to study completion in each study arm or between study arms (supplemental Table 2).
Efficacy
In the FAS, point estimates (95% confidence interval [CI]) of the proportions of PwHA with zero total, spontaneous, and spontaneous joint bleeds in the 1% to 3% vs 8% to 12% arms, respectively, were 42% (29% to 55%) vs 62% (49% to 75%; P = .055), 60% (47% to 72%) vs 76% (65% to 88%; P = .101), and 65% (53% to 77%) vs 85% (75% to 95%; P = .026) in the second 6 months (Table 2). Corresponding mean (SD) ABRs for total, spontaneous, and spontaneous joint bleeds were 3.6 (7.5) vs 1.6 (3.4), 2.5 (6.6) vs 0.7 (1.7), and 2.0 (6.4) vs 0.5 (1.7), respectively (Table 3). Prophylaxis with rurioctocog alfa pegol reduced total ABR during the 12-month study period in both treatment arms vs historical total ABR of PwHA in the 12 months before enrollment (supplemental Table 3). The primary outcome analysis in the FAS used multiple imputations for missing information on bleeding events in PwHA who did not have 364 days of observation (5/57 and 10/58 PwHA in the 1% to 3% and 8% to 12% arms, respectively). Of the cumulative planned observation time in each arm, 94/20 748 (0.5%) days in the 1% to 3% arm and 1611/21 112 (7.6%) days in the 8% to 12% arm did not have any available data because of early patient termination. In the PPAS, point estimates (95% CI) of the proportions of PwHA with zero total, spontaneous, and spontaneous joint bleeds in the 1% to 3% vs 8% to 12% arms, respectively, were 40% (27% to 55%) vs 67% (52% to 81%; P = .015), 60% (45% to 73%) vs 81% (67% to 92%; P = .038), and 65% (51% to 78%) vs 91% (78% to 97%; P = .008) in the second 6 months (Table 2). Corresponding mean (SD) ABRs for total, spontaneous, and spontaneous joint bleeds were lower in the 8% to 12% vs 1% to 3% arm (Table 3). Additional secondary efficacy outcomes are shown in Table 3 (joint ABR, ABR for joints with ≥4 spontaneous bleeds in 6 consecutive months, injury-related ABR) and supplemental Table 4 (nonjoint, major, moderate, minor ABRs).
Proportion of PwHA who had zero bleeds (second 6-mo study period; FAS and PPAS)
| FAS (N = 115) | PPAS (N = 95) | |||||
|---|---|---|---|---|---|---|
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 58) | P* | FVIII trough level 1% to 3% (n = 52) | FVIII trough level 8% to 12% (n = 43) | P* | |
| Zero total bleeds | ||||||
| Point estimate (95% CI) of proportion of PwHA, % | 42 (29-55) | 62 (49-75) | .055 | 40 (27-55) | 67 (52-81) | .015 |
| Zero spontaneous bleeds | ||||||
| Point estimate (95% CI) of proportion of PwHA, % | 60 (47-72) | 76 (65-88) | .101 | 60 (45-73) | 81 (67-92) | .038 |
| Zero spontaneous joint bleeds | ||||||
| Point estimate (95% CI) of proportion of PwHA, % | 65 (53-77) | 85 (75-95) | .026 | 65 (51-78) | 91 (78-97) | .008 |
| FAS (N = 115) | PPAS (N = 95) | |||||
|---|---|---|---|---|---|---|
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 58) | P* | FVIII trough level 1% to 3% (n = 52) | FVIII trough level 8% to 12% (n = 43) | P* | |
| Zero total bleeds | ||||||
| Point estimate (95% CI) of proportion of PwHA, % | 42 (29-55) | 62 (49-75) | .055 | 40 (27-55) | 67 (52-81) | .015 |
| Zero spontaneous bleeds | ||||||
| Point estimate (95% CI) of proportion of PwHA, % | 60 (47-72) | 76 (65-88) | .101 | 60 (45-73) | 81 (67-92) | .038 |
| Zero spontaneous joint bleeds | ||||||
| Point estimate (95% CI) of proportion of PwHA, % | 65 (53-77) | 85 (75-95) | .026 | 65 (51-78) | 91 (78-97) | .008 |
P < .05 between treatment arms was considered statistically significant.
Zero bleeds rate was based on intent-to-treat principle.
The null hypothesis of independence was tested against a 2-sided alternative by χ2 test with continuity correction.
ABRs (second 6-mo study period; FAS and PPAS)
| FAS (N = 115) | PPAS (N = 95) | |||
|---|---|---|---|---|
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 53)* | FVIII trough level 1% to 3% (n = 52) | FVIII trough level 8% to 12% (n = 43) | |
| Total ABR | ||||
| Mean (SD) | 3.6 (7.5) | 1.6 (3.4) | 2.8 (3.0) | 1.2 (2.4) |
| Median (Q1 to Q3) | 2.0 (0.0-4.0) | 0.0 (0.0-2.0) | 2.0 (0.0-4.0) | 0.0 (0.0-2.0) |
| Spontaneous ABR | ||||
| Mean (SD) | 2.5 (6.6) | 0.7 (1.7) | 1.7 (2.5) | 0.6 (1.5) |
| Median (Q1 to Q3) | 0.0 (0.0-4.0) | 0.0 (0.0-0.0) | 0.0 (0.0-4.0) | 0.0 (0.0-0.0) |
| Spontaneous joint ABR | ||||
| Mean (SD) | 2.0 (6.4) | 0.5 (1.7) | 1.2 (2.0) | 0.4 (1.4) |
| Median (Q1 to Q3) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) |
| Joint ABR | ||||
| Mean (SD) | 2.6 (7.4) | 1.1 (2.6) | 1.8 (2.2) | 0.8 (2.3) |
| Median (Q1 to Q3) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) | 1.0 (0.0-3.0) | 0.0 (0.0-0.0) |
| ABR of joints with ≥4 spontaneous bleeds in 6 consecutive months | ||||
| Mean (SD) | 1.0 (6.8) | 0.4 (1.5) | 0.1 (0.6) | 0.2 (1.3) |
| Median (Q1 to Q3) | 0.0 (0.0-0.0) | 0.0 (0.0-0.0) | 0.0 (0.0-0.0) | 0.0 (0.0-0.0) |
| Injury-related ABR | ||||
| Mean (SD) | 1.1 (2.0) | 0.9 (2.6) | 1.1 (1.9) | 0.7 (1.7) |
| Median (Q1 to Q3) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) |
| FAS (N = 115) | PPAS (N = 95) | |||
|---|---|---|---|---|
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 53)* | FVIII trough level 1% to 3% (n = 52) | FVIII trough level 8% to 12% (n = 43) | |
| Total ABR | ||||
| Mean (SD) | 3.6 (7.5) | 1.6 (3.4) | 2.8 (3.0) | 1.2 (2.4) |
| Median (Q1 to Q3) | 2.0 (0.0-4.0) | 0.0 (0.0-2.0) | 2.0 (0.0-4.0) | 0.0 (0.0-2.0) |
| Spontaneous ABR | ||||
| Mean (SD) | 2.5 (6.6) | 0.7 (1.7) | 1.7 (2.5) | 0.6 (1.5) |
| Median (Q1 to Q3) | 0.0 (0.0-4.0) | 0.0 (0.0-0.0) | 0.0 (0.0-4.0) | 0.0 (0.0-0.0) |
| Spontaneous joint ABR | ||||
| Mean (SD) | 2.0 (6.4) | 0.5 (1.7) | 1.2 (2.0) | 0.4 (1.4) |
| Median (Q1 to Q3) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) |
| Joint ABR | ||||
| Mean (SD) | 2.6 (7.4) | 1.1 (2.6) | 1.8 (2.2) | 0.8 (2.3) |
| Median (Q1 to Q3) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) | 1.0 (0.0-3.0) | 0.0 (0.0-0.0) |
| ABR of joints with ≥4 spontaneous bleeds in 6 consecutive months | ||||
| Mean (SD) | 1.0 (6.8) | 0.4 (1.5) | 0.1 (0.6) | 0.2 (1.3) |
| Median (Q1 to Q3) | 0.0 (0.0-0.0) | 0.0 (0.0-0.0) | 0.0 (0.0-0.0) | 0.0 (0.0-0.0) |
| Injury-related ABR | ||||
| Mean (SD) | 1.1 (2.0) | 0.9 (2.6) | 1.1 (1.9) | 0.7 (1.7) |
| Median (Q1 to Q3) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) | 0.0 (0.0-2.0) | 0.0 (0.0-0.0) |
ABR based on 53 PwHA; data not available from 5 randomized PwHA who discontinued the study before the second 6-mo period.
In the post hoc analyses, change from baseline in the number of joints with ≥4 spontaneous bleeds was assessed in 110 of 115 PwHA who had ≥6 months of observation during the 12-month study period. The number of joints with ≥4 spontaneous bleeds decreased in all but 1 patient in the 1% to 3% arm (2 remaining joints with ≥4 spontaneous bleeds each) and 1 patient in the 8% to 12% arm (1 remaining joint with ≥4 spontaneous bleeds) after 6 months of rurioctocog alfa pegol prophylaxis (supplemental Table 5).
ABRs in the PPAS during the second 6 months were stratified by patients’ FVIII replacement regimens before enrollment (on-demand or prophylactic). A larger difference in ABRs (total, spontaneous, injury-related) between treatment arms was observed in PwHA who only received on-demand treatment before enrollment vs those who received prior prophylaxis (supplemental Table 6).
Rurioctocog alfa pegol consumption
Weight-adjusted prophylactic consumption of rurioctocog alfa pegol over the 12-month study period is summarized in Table 4. In the FAS, PwHA in the 1% to 3% and 8% to 12% arms, respectively, administered a median (Q1 to Q3) of 2.0 (2.0 to 2.3) and 3.4 (3.1 to 3.6) infusions per week, 30.3 (23.3 to 40.3) and 38.4 (26.8 to 51.6) IU/kg per infusion, and 66.2 (51.3 to 96.3) and 143.6 (91.4 to 189.8) IU/kg per week, with similar results observed in the PPAS.
Rurioctocog alfa pegol prophylaxis consumption (12-mo study period; FAS and PPAS)
| FAS (N = 115) | PPAS (N = 95) | |||
|---|---|---|---|---|
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 58) | FVIII trough level 1% to 3% (n = 52) | FVIII trough level 8% to 12% (n = 43) | |
| Number of prophylactic infusions per week | ||||
| Mean (SD) | 2.3 (0.6) | 3.6 (1.2) | 2.3 (0.6) | 3.8 (1.2) |
| Median (Q1 to Q3) | 2.0 (2.0-2.3) | 3.4 (3.1-3.6) | 2.0 (2.0-2.3) | 3.5 (3.3-4.3) |
| Range | 1.5-3.5 | 1.6-6.8 | 1.5-3.5 | 1.6-6.8 |
| Number of prophylactic infusions per year | ||||
| Mean (SD) | 117.9 (30.3) | 189.7 (61.6) | 117.8 (30.0) | 198.6 (64.7) |
| Median (Q1 to Q3) | 104.4 (102.3-121.4) | 179.4 (162.6-188.5) | 104.4 (102.3-121.4) | 181.6 (171.6-222.8) |
| Range | 80.3-181.6 | 83.3-356.2 | 80.3-181.6 | 83.3-356.2 |
| Prophylactic dose per infusion (IU/kg) | ||||
| Mean (SD) | 33.0 (12.6) | 40.3 (14.2) | 33.6 (13.0) | 37.7 (13.3) |
| Median (Q1 to Q3) | 30.3 (23.3-40.3) | 38.4 (26.8-51.6) | 31.7 (23.1-41.7) | 34.2 (26.1-48.4) |
| Range | 15.2-65.2 | 16.2-73.6 | 15.2-65.2 | 18.8-73.6 |
| Prophylactic dose per week (IU/kg) | ||||
| Mean (SD) | 74.0 (31.8) | 143.3 (56.2) | 75.3 (32.5) | 142.4 (60.0) |
| Median (Q1 to Q3) | 66.2 (51.3-96.3) | 143.6 (91.4-189.8) | 67.0 (51.5-96.6) | 140.2 (89.3-191.1) |
| Range | 30.9-163.6 | 39.0-253.3 | 30.9-163.6 | 39.0-253.3 |
| Prophylactic dose per year (IU/kg) | ||||
| Mean (SD) | 3860.8 (1658.0) | 7478.2 (2934.5) | 3930.0 (1695.0) | 7432.6 (3127.8) |
| Median (Q1 to Q3) | 3453.9 (2678.9-5022.4) | 7490.2 (4771.6-9903.3) | 3497.2 (2688.9-5039.5) | 7316.2 (4659.7-9972.6) |
| Range | 1614.3-8535.3 | 2036.3-13 215.5 | 1614.3-8535.3 | 2036.3-13 215.5 |
| FAS (N = 115) | PPAS (N = 95) | |||
|---|---|---|---|---|
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 58) | FVIII trough level 1% to 3% (n = 52) | FVIII trough level 8% to 12% (n = 43) | |
| Number of prophylactic infusions per week | ||||
| Mean (SD) | 2.3 (0.6) | 3.6 (1.2) | 2.3 (0.6) | 3.8 (1.2) |
| Median (Q1 to Q3) | 2.0 (2.0-2.3) | 3.4 (3.1-3.6) | 2.0 (2.0-2.3) | 3.5 (3.3-4.3) |
| Range | 1.5-3.5 | 1.6-6.8 | 1.5-3.5 | 1.6-6.8 |
| Number of prophylactic infusions per year | ||||
| Mean (SD) | 117.9 (30.3) | 189.7 (61.6) | 117.8 (30.0) | 198.6 (64.7) |
| Median (Q1 to Q3) | 104.4 (102.3-121.4) | 179.4 (162.6-188.5) | 104.4 (102.3-121.4) | 181.6 (171.6-222.8) |
| Range | 80.3-181.6 | 83.3-356.2 | 80.3-181.6 | 83.3-356.2 |
| Prophylactic dose per infusion (IU/kg) | ||||
| Mean (SD) | 33.0 (12.6) | 40.3 (14.2) | 33.6 (13.0) | 37.7 (13.3) |
| Median (Q1 to Q3) | 30.3 (23.3-40.3) | 38.4 (26.8-51.6) | 31.7 (23.1-41.7) | 34.2 (26.1-48.4) |
| Range | 15.2-65.2 | 16.2-73.6 | 15.2-65.2 | 18.8-73.6 |
| Prophylactic dose per week (IU/kg) | ||||
| Mean (SD) | 74.0 (31.8) | 143.3 (56.2) | 75.3 (32.5) | 142.4 (60.0) |
| Median (Q1 to Q3) | 66.2 (51.3-96.3) | 143.6 (91.4-189.8) | 67.0 (51.5-96.6) | 140.2 (89.3-191.1) |
| Range | 30.9-163.6 | 39.0-253.3 | 30.9-163.6 | 39.0-253.3 |
| Prophylactic dose per year (IU/kg) | ||||
| Mean (SD) | 3860.8 (1658.0) | 7478.2 (2934.5) | 3930.0 (1695.0) | 7432.6 (3127.8) |
| Median (Q1 to Q3) | 3453.9 (2678.9-5022.4) | 7490.2 (4771.6-9903.3) | 3497.2 (2688.9-5039.5) | 7316.2 (4659.7-9972.6) |
| Range | 1614.3-8535.3 | 2036.3-13 215.5 | 1614.3-8535.3 | 2036.3-13 215.5 |
Treatment of a breakthrough bleed in the 1% to 3% and 8% to 12% arms required a similar number of infusions and dose per infusion in the FAS and PPAS (supplemental Table 7).
Adherence to prophylaxis
Adherence to individualized prophylaxis schedules was observed in 86.0% (49/57) of PwHA in the 1% to 3% arm and 93.1% (54/58) in the 8% to 12% arm of the FAS. Adherence to required dose was observed in 86.0% (49/57) of PwHA in the 1% to 3% arm and 75.9% (44/58) in the 8% to 12% arm.
Safety and immunogenicity
The SAS comprised 115 randomized PwHA who received ≥1 infusion of rurioctocog alfa pegol. Overall, 204 AEs (nonserious and serious) occurred in 60.9% (70/115) of PwHA (excluding surgery periods; Table 5). All AEs are reported in supplemental Table 8. No AEs resulted in discontinuation, and no deaths, serious thrombotic events, or severe allergic reactions were observed. A total of 9 serious AEs were reported in 7 PwHA (Table 5). One patient in the 8% to 12% arm had a serious AE considered by the investigator and sponsor to be related to rurioctocog alfa pegol. This patient had a positive FVIII inhibitor test of 0.6 BU (week 8) according to central laboratory testing (Bethesda assay cutoff of ≥0.6 BU), and FVIII inhibitors resolved within 18 days and remained negative (≤0.4 BU) until study completion. The patient did not have any symptoms of inhibitor development, and FVIII kinetics were not affected; anti-FVIII binding antibodies were negative throughout the study.
AEs in PwHA who received ≥1 prophylactic rurioctocog alfa pegol dose (randomized PwHA in SAS*)
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 58) | All PwHA (N = 115) | |
|---|---|---|---|
| Nonserious and serious AEs* | |||
| All AEs, n | 101 | 103 | 204 |
| PwHA with AEs, n (%) | 34 (59.6) | 36 (62.1) | 70 (60.9) |
| AEs considered related to rurioctocog alfa pegol,† n | 2 | 2 | 4 |
| Nonserious AEs | |||
| All nonserious AEs, n | 97 | 98 | 195 |
| PwHA with nonserious AEs, n (%) | 32 (56.1) | 35 (60.3) | 67 (58.3) |
| Serious AEs | |||
| All serious AEs, n | 4 | 5 | 9‡ |
| PwHA with serious AEs, n (%) | 3 (5.3) | 4 (6.9) | 7 (6.1) |
| Serious AEs considered related to rurioctocog alfa pegol,† n | 0 | 1 | 1 |
| Anti-FVIII inhibitory antibodies, n | 0 | 1§ | 1§ |
| Death, n | 0 | 0 | 0 |
| FVIII trough level 1% to 3% (n = 57) | FVIII trough level 8% to 12% (n = 58) | All PwHA (N = 115) | |
|---|---|---|---|
| Nonserious and serious AEs* | |||
| All AEs, n | 101 | 103 | 204 |
| PwHA with AEs, n (%) | 34 (59.6) | 36 (62.1) | 70 (60.9) |
| AEs considered related to rurioctocog alfa pegol,† n | 2 | 2 | 4 |
| Nonserious AEs | |||
| All nonserious AEs, n | 97 | 98 | 195 |
| PwHA with nonserious AEs, n (%) | 32 (56.1) | 35 (60.3) | 67 (58.3) |
| Serious AEs | |||
| All serious AEs, n | 4 | 5 | 9‡ |
| PwHA with serious AEs, n (%) | 3 (5.3) | 4 (6.9) | 7 (6.1) |
| Serious AEs considered related to rurioctocog alfa pegol,† n | 0 | 1 | 1 |
| Anti-FVIII inhibitory antibodies, n | 0 | 1§ | 1§ |
| Death, n | 0 | 0 | 0 |
AEs that occurred after the first randomized dose was received until study end; excludes any AEs that occurred during the surgical period.
Per investigator and sponsor assessment.
Four serious AEs in 3 PwHA with 1% to 3% FVIII trough levels (moderate severity: 1 limb abscess, 1 event of cellulitis, 1 event of appendicitis, 1 radius fracture) and 5 serious AEs in 4 PwHA with 8% to 12% FVIII trough levels (moderate severity: 1 hand fracture, 1 head injury, 1 event of multiple injuries; mildly serious AEs: 1 laceration, 1 event of FVIII inhibition).
Transient low-titer anti-FVIII inhibitory antibodies (0.6 BU at week 8) that resolved before study end. Anti-FVIII binding antibodies for this patient were negative throughout the study.
In the 1% to 3% arm, 3 PwHA had single positive results for binding antibodies at screening/baseline only (anti–PEG-FVIII immunoglobulin G [IgG] and anti–PEG IgM). In the 8% to 12% arm, 9 PwHA had binding antibodies at baseline that were also detectable during the study. Eight of these PwHA in the 8% to 12% arm had transient expression of binding antibodies (anti-FVIII IgG, anti–PEG-FVIII IgG, or anti–PEG-FVIII IgM), and 1 patient was positive for anti–PEG-FVIII IgG from screening through study completion, including 1 negative result. Binding antibodies that were detected could not be correlated to impaired treatment efficacy or related AEs. In 1 patient, binding antibodies were associated with decreased recovery during a PK assessment vs baseline recovery (before development of binding antibodies), although recovery remained within normal range.
PK
At the initial PK assessment in randomized PwHA in the 1% to 3% and 8% to 12% arms, respectively, rurioctocog alfa pegol had a mean (SD) t1/2 of 15.3 (4.2) and 14.7 (5.1) hours. Mean (SD) IR levels at maximum plasma concentration were similar in the 1% to 3% and 8% to 12% arms, respectively (2.2 [0.5] and 2.2 [0.6] IU/dL / IU/kg).
Overall exposure to FVIII was based on average FVIII activity levels over time. In the 1% to 3% and 8% to 12% arms, respectively, FVIII activity was a median (Q1 to Q3) 17.3 (15.2 to 21.7) and 35.0 (29.2 to 40.9) IU/dL during the first 6 months, and 17.3 (14.5 to 22.4) and 30.9 (24.9 to 41.2) IU/dL during the second 6 months (supplemental Table 9).
Observed FVIII activity trough levels during the second 6 months were within the intended ranges of 1% to 3% and 8% to 12%; median FVIII troughs ranged from 2.1 to 3.0 IU/dL and 10.7 to 11.7 IU/dL, respectively. Box plots of calculated FVIII activity trough levels over time by treatment arm from week 4 to study completion are shown in supplemental Figure 1.
Discussion
Targeting FVIII trough levels ≥1% with a twice-weekly fixed-dose regimen of 40 to 50 IU/kg rurioctocog alfa pegol in PwHA with severe disease demonstrated comparable efficacy and safety profiles in previous phase 2 and 3 trials.11-13 However, data were lacking on potential benefits of targeting higher FVIII troughs with PK-guided prophylaxis in severe hemophilia A. PROPEL is the only prospective randomized clinical trial to date to compare safety and efficacy of PK-guided treatment targeting FVIII trough levels of 1% to 3% and 8% to 12% in severe hemophilia A. In this trial, intended FVIII troughs were reliably achieved and maintained during the second 6-month study period. PK-guided rurioctocog alfa pegol prophylaxis decreased ABRs in each treatment arm vs preenrollment historical ABRs. A higher proportion of PwHA had zero total bleeds with prophylaxis targeting FVIII troughs of 8% to 12% than 1% to 3%. This difference in favor of the 8% to 12% target FVIII trough level was considered clinically relevant but not statistically significant in the FAS (62% vs 42%; P = .055; primary endpoint). In the PPAS subgroup, consisting of PwHA who adhered to their intended time in the study, dose, and schedule, the difference was similarly favorable in magnitude and statistically significant (67% vs 40%; P = .015). The higher proportion of PwHA with zero bleeds in the 8% to 12% arm was also statistically significant for spontaneous ABR (81% vs 60%; P = .038) and spontaneous joint ABR (91% vs 65%; P = .008) in the PPAS.
Accordingly, consistently lower bleeding rates were observed in the 8% to 12% arm, with total ABR of <2 in the 8% to 12% arm and ≥2 in the 1% to 3% arm of the FAS. There were similar differences in ABR between treatment arms for spontaneous, spontaneous joint, joint, target joint, major bleeds, and injury-related bleeds not necessarily related to hemophilia A. Evaluation of multiple bleeds into the same joint did not follow the currently accepted definition of target joint by the International Society of Thrombosis and Hemostasis (≥3 spontaneous bleeds into a single joint within a consecutive 6-month study period)20 because development of the study protocol began before the International Society of Thrombosis and Hemostasis definition was published. Therefore, instead of assessing target joints, this study evaluated the presence of ≥4 spontaneous bleeds into a single joint in any consecutive 6-month period. The number of joints with ≥4 spontaneous bleeds decreased within the first 6 months of prophylaxis in all but 1 patient per study arm. These findings suggest that sustaining FVIII troughs of 1% to 3% may be sufficient to prevent frequent spontaneous bleeds into the same joint, but 8% to 12% FVIII troughs are needed to prevent all bleeds in a higher proportion of PwHA.
AEs reported in both treatment arms were comparable and were consistent with previous rurioctocog alfa pegol trials.11-13 One patient exhibited transient positive FVIII inhibitory activity related to rurioctocog alfa pegol without evidence of a binding anti-FVIII antibody; FVIII inhibitors resolved in 18 days without specific treatment. The investigator and sponsor concluded that development of this putative transient inhibitor had no impact on FVIII PK, as constant FVIII troughs were observed over the entire study period.
Many PwHA met their target FVIII troughs using rurioctocog alfa pegol prophylaxis at rates within or below the recommended weekly dose (40 to 50 IU/kg, twice weekly9 ). Overall, weekly consumption was variable, and overlapping ranges between treatment arms likely reflect the heterogeneity of patients’ FVIII t1/2. This finding emphasizes that personalized treatment should be considered based on the patient’s PK profile and pattern of bleeding.21 Targeting an elevated FVIII trough level may benefit PwHA who can achieve higher troughs with a reasonable dose consumption and reasonable injection frequency. Dosing intervals used to achieve 8% to 12% FVIII troughs were mostly the accepted standard FVIII every-other-day and extended regimens. Of note, only 1 patient in the 8% to 12% arm discontinued treatment owing to daily injections. Targeting an elevated FVIII trough may also benefit PwHA with continued bleeds despite ongoing standard prophylaxis, and those seeking more active lifestyles.22 In addition, higher FVIII peak levels and area under the FVIII-level curve were previously suggested to contribute to bleed protection.22 In PROPEL, a higher overall exposure to FVIII was achieved in the 8% to 12% vs 1% to 3% arm during the full study period, potentially contributing to the observed benefit in this population.
There are a few limitations to this study. The study population comprised adolescents and adults with rather high historical ABR ≥2, including PwHA already receiving prophylaxis; therefore, results may only be generalizable to PwHA with a history of moderate to high ABRs. The proportion of PwHA with ≥4 joints with ≥4 spontaneous bleeds or arthropathy at screening was approximately twice as great in the 8% to 12% vs 1% to 3% arm; however, baseline demographics and other disease characteristics were similar between treatment arms. The higher discontinuation rate from the 8% to 12% vs 1% to 3% arm and greater number of PwHA excluded from the PPAS in the 8% to 12% arm were limitations of the interpretation of the data obtained in this study. Although a relationship between these findings and burden of targeting 8% to 12% FVIII troughs cannot be ruled out, the primary endpoint analysis in the FAS accounted for discontinuations, and despite higher exposure to FVIII in the 8% to 12% arm, none of the discontinuations were related to rurioctocog alfa pegol safety. In addition, a sensitivity analysis that assumed more bleeds than predicted from observed data demonstrated only a small impact on the proportion of PwHA with zero total bleeds in 6 months in the 8% to 12% arm (data not shown). The 6 months of follow-up could be considered a limitation, as 12 months of follow-up would provide more opportunity to observe the difference in bleeding rates between treatment arms. In conclusion, PK-guided prophylaxis with rurioctocog alfa pegol that targeted FVIII trough levels of 1% to 3% or 8% to 12% was feasible and efficacious, and targeting 8% to 12% FVIII troughs resulted in a higher proportion of PwHA with no bleeds than prophylaxis that targeted 1% to 3% FVIII troughs. These results support the hypothesis that an elevated FVIII trough can benefit PwHA without changing the safety profile. Overall, findings from this study emphasize that personalized treatment of PwHA should be considered.
The data sets, including the redacted study protocol, redacted statistical analysis plan, and individual participants’ data supporting the results reported in this article, will be made available within 3 months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. Data requests should follow the process outlined in the Data Sharing section on: www.takeda.com/what-we-do/research-and-development/takeda-clinical-trial-transparency/. The study sponsor analyzed the data and conducted the statistical analyses. Data were available to all authors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all the patients and their caregivers who took part in the PROPEL study, as well as the PROPEL study investigators and sites (#NCT02585960; https://clinicaltrials.gov/ct2/show/NCT02585960).
Under the direction of the authors, medical writing support was provided by Daniella A. Babu, employee of Excel Medical Affairs (Fairfield, CT), and was funded by Baxalta US Inc, a Takeda company (Lexington, MA). This study was supported by research funding from Baxalta US Inc, a Takeda company, and Baxalta Innovations GmbH, a Takeda company (Vienna, Austria). W.S. was an employee of Baxalta US Inc, a Takeda company, during the analysis of this study.
Authorship
Contribution: R.K., J.W., V.R., P.W.C., O.S., and H.M.I. contributed to the interpretation of the data and study conduct; W.E. and B.E. contributed to the conception and design of the study, and to the analysis and interpretation of the data; S.D.T. and W.S. contributed to the analysis and interpretation of the data, and study conduct; and all authors revised the manuscript critically for intellectual content and gave their final approval for it to be published.
Conflict-of-interest disclosure: R.K. has received grant/research support from Bayer, BioMarin, CSL Behring, Novo Nordisk, Octapharma, Pfizer, and Shire (a Takeda company); consultancy fees from Bayer, BioMarin, Biotest, CSL Behring, Grifols, Novo Nordisk, Octapharma, Pfizer, Roche, Shire (a Takeda company), and Sobi; and has served on speakers bureaus for Bayer, BioMarin, Biotest, CSL Behring, Grifols, Novo Nordisk, Octapharma, Pfizer, Roche, Shire (a Takeda company), and Sobi. J.W. has received grant/research support and honoraria for lectures from Alexion, Alnylam, Baxalta/Shire (a Takeda company), Bayer, CSL Behring, Ferring, Novo Nordisk, Octapharma, Rigel, Roche, Sanofi, Siemens, Sobi, and Werfen. V.R. has received research support from Grifols, Pfizer, and Takeda. P.W.C. has received research support from CSL Behring; consultancy fees from CSL Behring, Novo Nordisk, Shire (a Takeda company), and Sobi; and has served on a speakers bureau for Shire (a Takeda company). O.S. has received honoraria from CSL Behring, Novo Nordisk, and Shire (a Takeda company); and has served on speakers bureaus for Novo Nordisk, Pfizer, and Shire (a Takeda company). H.M.I. has received grant/research support from Novartis, Sanofi, and Shire (a Takeda company). W.E. is an employee of Baxalta Innovations GmbH, a Takeda company, and a Takeda stock owner. S.D.T. and B.E. are employees of Baxalta US Inc, a Takeda company, and Takeda stock owners. W.S. was an employee of Baxalta US Inc, a Takeda company, during the analysis of this study.
Correspondence: Robert Klamroth, Vivantes Klinikum im Friedrichshain, Landsberger Allee 49, 10249 Berlin, Germany; e-mail: robert.klamroth@vivantes.de.
