

Key Points
A zinc transporter, TMEM163, is essential for the biogenesis and function of platelet DGs.
BLOC-1 complex is involved in the trafficking of TMEM163 to platelet DGs, which explains the defective DGs in HPS.

Abstract
Lysosome-related organelles (LROs) are a category of secretory organelles enriched with ions such as calcium, which are maintained by ion transporters or channels. Homeostasis of these ions is important for LRO biogenesis and secretion. Hermansky-Pudlak syndrome (HPS) is a recessive disorder with defects in multiple LROs, typically platelet dense granules (DGs) and melanosomes. However, the underlying mechanism of DG deficiency is largely unknown. Using quantitative proteomics, we identified a previously unreported platelet zinc transporter, transmembrane protein 163 (TMEM163), which was significantly reduced in BLOC-1 (Dtnbp1sdy and Pldnpa)–, BLOC-2 (Hps6ru)–, or AP-3 (Ap3b1pe)–deficient mice and HPS patients (HPS2, HPS3, HPS5, HPS6, or HPS9). We observed similar platelet DG defects and higher intracellular zinc accumulation in platelets of mice deficient in either TMEM163 or dysbindin (a BLOC-1 subunit). In addition, we discovered that BLOC-1 was required for the trafficking of TMEM163 to perinuclear DG and late endosome marker–positive compartments (likely DG precursors) in MEG-01 cells. Our results suggest that TMEM163 is critical for DG biogenesis and that BLOC-1 is required for the trafficking of TMEM163 to putative DG precursors. These new findings suggest that loss of TMEM163 function results in disruption of intracellular zinc homeostasis and provide insights into the pathogenesis of HPS or platelet storage pool deficiency.
Introduction
Hermansky-Pudlak syndrome (HPS) is a recessive disorder featured by oculocutaneous albinism, prolonged bleeding, and ceroid deposition. It is also accompanied by lung fibrosis, colitis, and other complications in some cases.1,2 The characteristic hallmarks of HPS are defects in multiple lysosome-related organelles (LROs), including dense granules (DGs) in platelets, melanosomes in melanocytes, Weibel-Palade bodies in endothelial cells, large-dense core vesicles in adrenal chromaffin cells, synaptic vesicles (SVs) in neurons, and lamellar bodies in alveolar type II epithelial cells.2-6 LROs are regarded as a category of secretory organelles sharing properties with conventional lysosomes, such as highly glycosylated integral lysosome-associated membrane proteins (LAMPs) and enrichment of intralumenal ions such as calcium (Ca2+).2,3,7
As inferred from melanosome biogenesis studies, platelet DGs are thought to originate via intermediary endosomes and multivesicular bodies (MVBs), and proteins may be transported to immature or mature DGs by a variety of protein complexes, such as the HPS protein–associated complexes (HPACs).2,8-14 However, the underlying mechanisms whereby HPACs affect DG biogenesis remain uncharacterized. In addition, Rab32 and Rab38 regulate the transport of LAMP2 and VMAT2 (DG integral membrane protein) from early endosomes (EEs) to mepacrine positive compartments.10,15 The Rab32 and Rab38 double mutant mimics severe HPS and has decreased DGs.16 Rab32/Rab38 may not directly interact with these cargoes (eg, LAMP2 or VMAT2), and whether LAMP2 or VMAT2 deficiency causes defective DG biogenesis is unknown. Deficiency of SLC35D3 (a transmembrane protein) affects DG biogenesis,17,18 but there is no clear evidence that SLC35D3 is localized to DGs. Altogether, the underlying mechanisms of DG biogenesis are largely unknown.
Traditionally, ion channels play a pivotal role in LRO biogenesis. Two-pore channel 2 (TPC2) regulates the pH, release of Ca2+, membrane dynamics, and content exchange of mepacrine-positive compartments in MEG-01 cells.19 In melanocytes, TPC2 regulates melanosomal pH and size by mediating Ca2+ release from melanosomes, thus controlling pigmentation.20 Our recent studies demonstrated that mitochondrial NCKX5 (a member of the K+-dependent Na+/Ca2+ exchanger family) participates in melanosome maturation and pigment production by regulating Ca2+ homeostasis of melanosomes.21 Thus, similar to the melanosome, homeostasis of lumenal ions is important for DG biogenesis. However, knowledge of ion transporters in DGs is very limited.
Rats have an increased bleeding tendency upon maintenance on a Zn2+-deficient diet.22-24 In addition, a bleeding tendency along with abnormal platelet aggregation was noted in 2 cancer patients (one with squamous cell carcinoma and another with non-Hodgkin lymphoma) presenting with a nutritional Zn2+ deficiency. Normal bleeding times and platelet aggregation were restored by dietary zinc (Zn2+) supplementation.25 Zn2+ is known as a mediator of hemostasis.26-30 Upon incubation with exogenous Zn2+, platelets stained with the fluorescent Zn2+ indicator FluoZin-3 display a rapid fluorescence increase,31 indicating that platelet Zn2+ entry pathways exist. It has been reported that intracellular Zn2+ homeostasis is maintained by 2 highly conserved Zn2+ transporter protein families, ZIP (SLC39) and ZNT (SLC30), in mammalian cells. The 14 members of ZIP protein family regulate influx of Zn2+ into the cytoplasm from the extracellular matrix and the lumen of vesicular compartments. By contrast, the 10 members of ZNT protein family regulate efflux of Zn2+ into the extracellular matrix and the lumen of vesicular compartments from the cytoplasm.32-39 However, there is no report of Zn2+-selective ion channels or transporters in platelets. Zn2+ has been proposed as an intracellular second messenger, analogous to Ca2+, being released into the cytosol from intracellular storage pools.40 However, the storage pools of Zn2+ in platelets have not been resolved.
In rat brain synaptosomes, transmembrane protein 163 (TMEM163), also called synaptic vesicle 31 (SV31), was first identified as a SV membrane protein binding Zn2+.41 Exogenously expressed rodent TMEM163 mainly localized to the plasma membrane, lysosomes, and SVs.42-44 Purified rodent TMEM163 reconstituted in lipid bilayers assembles into a dimer and functions as a proton-dependent Zn2+ transporter.45 In addition, TMEM163 interacts with transient receptor potential mucolipin-1 (TRPML1), which has been implicated in mucolipidosis type IV (MLIV) pathogenesis. Both fibroblasts of a MLIV patient and human embryonic kidney (HEK293) cells with knockdown of TRPML1 showed abnormal lysosomal Zn2+ accumulation.43,44,46 Furthermore, TRPML1-deficient mice displayed increased Zn2+ levels in postmortem brain,46 suggesting that TRPML1 and TMEM163 play critical roles in intracellular Zn2+ homeostasis. In the Indian population, TMEM163 is a genome-wide association study candidate gene associated with insulin resistance and type 2 diabetes.47 Although, TMEM163 transcripts are detected in numerous tissues, including brain, kidney, lung, pancreas, testis, and ovary,41,43 the function of TMEM163 is controversial,43,48 and its presence, localization, and function in platelets are unknown.
In the present study, we find that TMEM163 is a critical component of the limiting membrane of the DG and late endosome (LE) marker–positive compartment (likely a precursor of DGs) in MEG-01 cells. TMEM163 is reduced in platelets of mice and HPS patients lacking HPACs (BLOC-1, BLOC-2 and AP-3). We demonstrate that BLOC-1 interacts with TMEM163 during its transport from EEs to perinuclear DG and LE marker–positive compartments. This may explain the similar DG defects in both Tmem163 knockout mice and some of the above-mentioned HPS mutant mice.
Materials and methods
Mice
The sandy (Dtnbp1sdy) mutant in the C57BL/6J background (wild-type [WT]) was originated from sdy mutant in the DBA/2J background49 through backcrossing at least 8 generations. Other mouse mutants and their controls, including pallid (Pldnpa), ruby-eye (Hps6ru), pale-ear (Hps1ep), pearl (Ap3b1pe), buff (Vps33abf), a double mutant of ruby-eye and pale-ear (ru/ep), and a double mutant of buff and pale-ear (bf/ep) in the C57BL/6J background, were derived as described previously.5,50 They were obtained from Richard T. Swank’s laboratory at Roswell Park Comprehensive Cancer Center (Buffalo, NY). The Tmem163 knockout mice (Tmem163-KO) were generated using Biosset’s EGE system based on CRISPR-Cas9 in the C57BL/6J background. To ensure the genotypes of the littermates, polymerase chain reaction (PCR) amplifications were designed according to the targeted deletion of exons 1 and 2. All mice in this study were bred in the animal facility of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences. All procedures were approved by the Institutional Animal Care and Use Committee of the Institute of Genetics and Developmental Biology. Age-matched WT mice (C57BL/6J, B6) or heterozygous mice were used as background controls.
Human subjects
The 12 HPS1 to HPS6 patients (3 HPS1, 1 HPS2, 2 HPS3, 1 HPS4, 2 HPS5, and 3 HPS6) were identified in our group, and their clinical features and genotypes were described previously.51 One HPS9 patient was identified recently.52 Their parents were recruited in this study as controls. This study was approved by the internal review board of the bioethics committees of Beijing Tongren Hospital and Beijing Children’s Hospital, Capital Medical University. The study followed the Declaration of Helsinki. Informed consent was signed by all subjects participating in this study. Peripheral blood samples (2 mL) were collected from all subjects for enrichment of platelets for immunoblotting.
Data analysis
All data were obtained from ≥3 independent experiments and expressed as mean ± standard error of the mean (SEM). All quantitative results were plotted by 1-way analysis of variance with Tukey’s posttest, except where specified otherwise (eg, Student t test). Error bars indicate SEM (*P < .05; **P < .01; ***P < .001; N.S., not significant). All graphs were drawn in GraphPad Prism 5 (La Jolla, CA).
Details of additional materials and methods can be found in supplemental Data (available on the Blood Web site)
Results
TMEM163 expression in platelets is reduced in murine mutants and human patients of the HPS protein–associated complexes BLOC-1, BLOC-2, and AP-3
To investigate how HPACs function in DG biogenesis, we isolated the platelets of sandy (sdy; deficiency in BLOC-1 subunit dysbindin) mice and their heterozygous littermates (sdy+/−) for mass spectrometry. Using quantitative proteomics, we identified an uncharacterized transmembrane protein, TMEM163 (SV31), that was decreased in sdy mice compared with sdy+/− mice. The reduction of this protein is similar to CD63 (a type of DG integral membrane protein) (Figure 1A-B; supplemental Table 1; supplemental Figure 1). To verify the mass spectrometry results, we quantified endogenous TMEM163 protein expression in platelet lysates from sdy and sdy+/− mice by immunoblotting (Figure 1C). For comparison, we measured other lysates of platelets from BLOC-1–deficient pallid (pa) (Figure 1D), BLOC-2-deficient ruby-eye (ru) (Figure 1E), AP-3-deficient pearl (pe) (Figure 1F), and double mutant of ruby-eye and pale-ear (ru/ep) (Figure 1I) mice and their littermate controls. BLOC-1, BLOC-2, and AP-3 mutants showed significant reduction of TMEM163 relative to littermate control mice (Figure 1K). Interestingly, reduced TMEM163 in BLOC-1–, BLOC-2–, and AP-3–deficient platelets is similar to observations of reduced SLC35D3 in these mutant mice,17 suggesting that both TMEM163 and SLC35D3 are likely transported by these HPACs. However, lysates of BLOC-3–deficient pale-ear (ep) (Figure 1G), buff (bf) with the VPS33AD251E mutation (Figure 1H), and double mutant of buff and pale-ear (bf/ep) (Figure 1J) mice showed no alteration of TMEM163 expression (Figure 1K). Consistently, 1 BLOC-1–deficient patient (1 HPS9), 7 BLOC-2–deficient patients (2 HPS3, 2 HPS5, and 3 HPS6), and 1 AP-3–deficient patient (1 HPS2) showed significant reductions of TMEM163 relative to healthy controls (Figure 1L-N). However, 4 BLOC-3–deficient patients (3 HPS1 and 1 HPS4) showed no alteration of TMEM163 expression (Figure 1M-N). These results agree with the finding that BLOC-1, BLOC-2, and AP-3 regulate cargo transport from endosomes to melanosomes in the same cargo-transport pathways in melanocytes,11,12,50,53-56 whereas BLOC-3 and HOPS are involved in different routes in LRO biogenesis.57 Altogether, these results indicate that TMEM163 may function in platelets and that BLOC-1, BLOC-2, and AP-3 may regulate the transport of TMEM163 to platelet LROs.
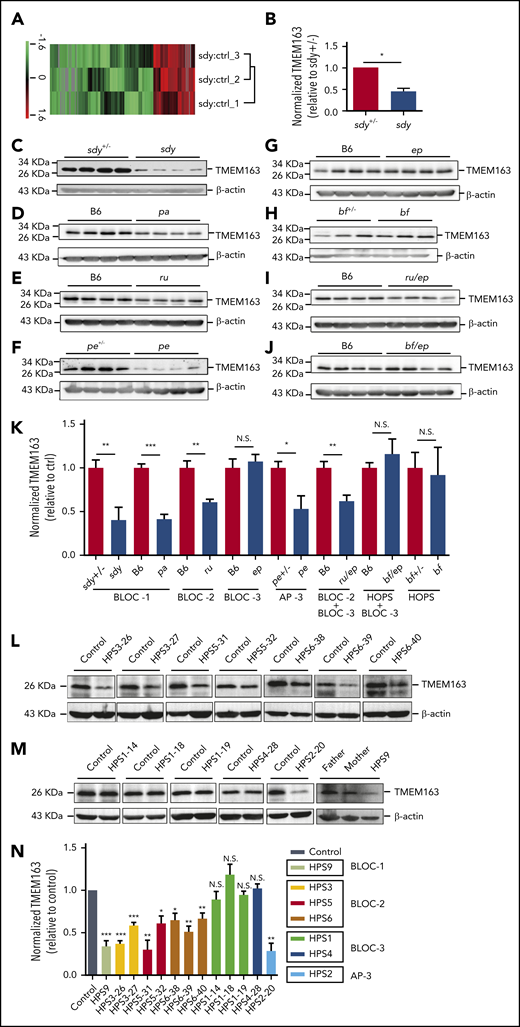
TMEM163 is reduced in platelets from HPS mouse models and patients with deficiency of BLOC-1, BLOC-2, or AP-3, but not BLOC-3. (A-B) Quantitative protein identification using liquid chromatography-mass spectrometery (LC-MS) of whole-platelet lysates in sdy mice compared with sdy+/− mice (ctrl). A total of 195 proteins (listed in supplemental Table 1) were selected using the Student t test and quantified with high confidence (P < .05). Hierarchy clustering of the z-scored isobaric tag for relative and absolute quantitation (iTRAQ) ratio of sdy to sdy+/− was used to evaluate the reproducibility of the experiments (A). The expression of TMEM163 in the platelets of sdy mice showed significant reduction compared with sdy+/− mice (B). n = 3, *P < .05. (C-J) Immunoblotting analysis of TMEM163 in platelets isolated from control mice and congenic HPS mutant mouse platelets with antibodies to TMEM163 or actin (a loading control). The expression of TMEM163 in platelets of sdy (BLOC-1-deficient HPS7 mutant), pa (BLOC-1-deficient HPS9 mutant), ru (BLOC-2-deficient HPS6 mutant), pe (AP-3-deficient HPS2 mutant), and ru/ep (BLOC-3-deficient model) double-mutant mice (both BLOC-2 and BLOC-3-deficient model) are significantly reduced compared with control mice (C-F,I). The expression of TMEM163 in platelets of ep, bf (the VPS33AD251E mutation model), and bf/ep showed no significant change compared with control mice (G-H,J). (K) TMEM163 band intensities were measured from ≥4 pairs of mice, normalized to actin levels, and expressed as the percentage (mean ± SEM) of the mean normalized value for control mice. (L) Immunoblotting assays showed the reduced TMEM163 in BLOC-2–deficient patients (2 HPS3, 2 HPS5, and 3 HPS6) compared with the healthy control (father or mother of each patient). (M) Immunoblotting assays showed normal TMEM163 in BLOC-3–deficient patients (3 HPS1 and 1 HPS4) and reduced TMEM163 in an AP-3–deficient patient (1 HPS2) and BLOC-1 deficient patient (1 HPS9) compared with the healthy control (father or mother of each patient). (N) Quantitative analysis of the TMEM163 band intensities. Each bar represents the percentage (mean ± SEM) of the mean normalized value for healthy control from 3 independent experiments. *P < .05; **P < .01; ***P < .001. ctrl, control; N.S., not significant.
TMEM163 is reduced in platelets from HPS mouse models and patients with deficiency of BLOC-1, BLOC-2, or AP-3, but not BLOC-3. (A-B) Quantitative protein identification using liquid chromatography-mass spectrometery (LC-MS) of whole-platelet lysates in sdy mice compared with sdy+/− mice (ctrl). A total of 195 proteins (listed in supplemental Table 1) were selected using the Student t test and quantified with high confidence (P < .05). Hierarchy clustering of the z-scored isobaric tag for relative and absolute quantitation (iTRAQ) ratio of sdy to sdy+/− was used to evaluate the reproducibility of the experiments (A). The expression of TMEM163 in the platelets of sdy mice showed significant reduction compared with sdy+/− mice (B). n = 3, *P < .05. (C-J) Immunoblotting analysis of TMEM163 in platelets isolated from control mice and congenic HPS mutant mouse platelets with antibodies to TMEM163 or actin (a loading control). The expression of TMEM163 in platelets of sdy (BLOC-1-deficient HPS7 mutant), pa (BLOC-1-deficient HPS9 mutant), ru (BLOC-2-deficient HPS6 mutant), pe (AP-3-deficient HPS2 mutant), and ru/ep (BLOC-3-deficient model) double-mutant mice (both BLOC-2 and BLOC-3-deficient model) are significantly reduced compared with control mice (C-F,I). The expression of TMEM163 in platelets of ep, bf (the VPS33AD251E mutation model), and bf/ep showed no significant change compared with control mice (G-H,J). (K) TMEM163 band intensities were measured from ≥4 pairs of mice, normalized to actin levels, and expressed as the percentage (mean ± SEM) of the mean normalized value for control mice. (L) Immunoblotting assays showed the reduced TMEM163 in BLOC-2–deficient patients (2 HPS3, 2 HPS5, and 3 HPS6) compared with the healthy control (father or mother of each patient). (M) Immunoblotting assays showed normal TMEM163 in BLOC-3–deficient patients (3 HPS1 and 1 HPS4) and reduced TMEM163 in an AP-3–deficient patient (1 HPS2) and BLOC-1 deficient patient (1 HPS9) compared with the healthy control (father or mother of each patient). (N) Quantitative analysis of the TMEM163 band intensities. Each bar represents the percentage (mean ± SEM) of the mean normalized value for healthy control from 3 independent experiments. *P < .05; **P < .01; ***P < .001. ctrl, control; N.S., not significant.
TMEM163 localizes to perinuclear DG and LE marker–positive compartments and EE-like organelles
Platelet α granules (AGs) and DGs are 2 major types of intracellular organelles that belong to the LRO family.9 Due to lack of an applicable antibody for endogenous TMEM163 immunofluorescence staining, we used a human megakaryocytic cell line, MEG-01, to determine the localization of TMEM163.8,10 MEG-01 cells were simultaneously transfected with different tagged-TMEM163 constructs (supplemental Figure 2), and we monitored Pearson correlation coefficients (PCCs) for the colocalization with multiple organelle markers. Confocal fluorescence microscopy revealed a high proportion of TMEM163 colocalization with platelet DG markers (Cherry-VMAT2, GFP-VMAT2, mepacrine, and CD63) (PCCs, 0.74 ± 0.04, 0.76 ± 0.02, 0.68 ± 0.04, and 0.73 ± 0.03, respectively) (Figure 2A-D,M), an LE marker (Rab7) (PCC, 0.72 ± 0.03) (Figure 2E), a lysosome marker (Lamp1) (PCC, 0.75 ± 0.02) (Figure 2F), and TPCN2 (likely in DG, LE or lysosome) (PCC, 0.59 ± 0.04) (Figure 2G). Consistently, live MEG-01 cells expressing Cherry-TMEM163 showed a high proportion of TMEM163 colocalization with LysoSensor Green DND-189 (PCC, 0.82 ± 0.02) (Figure 2H).58 Therefore, TMEM163 is concentrated in acidic perinuclear DG and LE marker–positive compartments. Further analysis showed epitope-tagged TMEM163 overlapped with EE markers, EEA1 (PCC, 0.40 ± 0.02) (Figure 2I) and Rab5 (PCC, 0.63 ± 0.03) (Figure 2J). The DG positive limiting membrane protein VMAT2 overlapped to only a small proportion with EEA1 (PCC, 0.14 ± 0.01) (supplemental Figure 3B) compared with mepacrine (PCC, 0.81 ± 0.02) (supplemental Figure 3A). Notably, TMEM163 showed very little colocalization with AG markers (P-selectin, von Willebrand factor [VWF], and PF4) (PCCs, 0.13 ± 0.02, 0.04 ± 0.01, and 0.08 ± 0.01, respectively) (Figure 2K-L; supplemental Figure 3C-D). Furthermore, sucrose density gradient centrifugation assays (Figure 2N) showed that TMEM163 fractionated in F3-F9. TMEM163 partially cofractionated with the EE marker EEA1 in F3 to F5, the LE marker Rab7 in F6 to F9, and the DG marker LAMP2 in F5 to F8, further indicating that TMEM163 is localized to endosomal structures. Together, these results suggest that TMEM163 localized mostly to perinuclear DG and LE marker–positive compartments (likely DG precursors) and small vesicular structures (likely EEs) in the periphery of MEG-01 cells. The localization of TMEM163 in EE-like and DG precursor-like compartments suggests that TMEM163 may traffic from EEs to DGs.
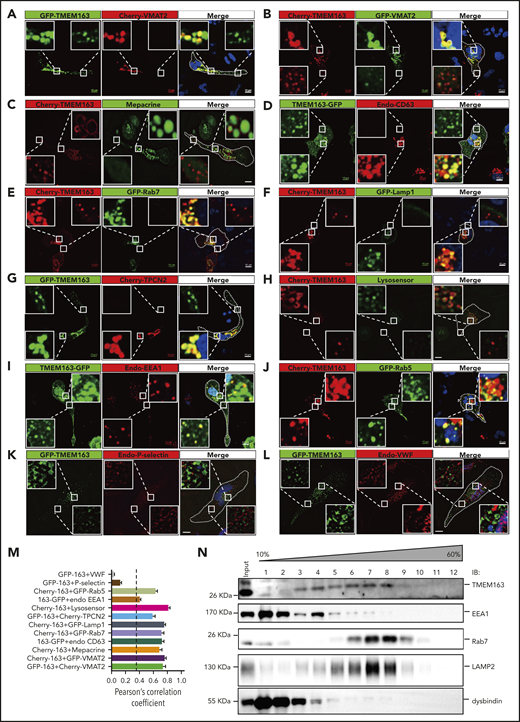
TMEM163 is localized to perinuclear DG and LE marker–positive compartments (likely DG precursors) and small vesicular structures (likely EEs) in the periphery, but not to AGs in MEG-01 cells. (A-B) MEG-01 cells were cotransfected with TMEM163-GFP or Cherry-TMEM163 and Cherry-VMAT2 (A) or GFP-VMAT2 (B) for 48 hours, respectively. (C) MEG-01 cells were transfected with Cherry-TMEM163 for 48 hours and then treated with 50 μM mepacrine for 30 minutes at 37°C. (D) MEG-01 cells were transfected with TMEM163-GFP for 48 hours and then fixed and stained with antibodies against CD63. (E-F) MEG-01 cells were cotransfected with Cherry-TMEM163 and GFP-Rab7 (E) or GFP-Lamp1 (F) for 48 hours, respectively. (G) MEG-01 cells were cotransfected with GFP-TMEM163 and Cherry-TPCN2 for 48 hours, respectively. (H) MEG-01 cells were transfected with Cherry-TMEM163 for 48 hours and treated with LysoSensor Green DND-189 for 30 minutes at 37°C. (I) MEG-01 cells were transfected with TMEM163-GFP for 48 hours and then fixed and stained with antibodies against EEA1. (J) MEG-01 cells were cotransfected with Cherry-TMEM163 and GFP-Rab5 for 48 hours. (K-L) MEG-01 cells were transfected with GFP-TMEM163 for 48 hours and then fixed and stained with antibodies against P-selectin (K) or VWF (L). Pictures shown in panels A-L are representative confocal images. Outlines of cells are indicated by white lines. Insets show 5× magnified images of the boxed region. Scale bars, 10 μm. (M) Colocalization analysis of results shown in panels A-L; PCC, 0.74 ± 0.04, 0.76 ± 0.02, 0.68 ± 0.04, 0.73 ± 0.03, 0.72 ± 0.03, 0.75 ± 0.02, 0.59 ± 0.04, 0.82 ± 0.02, 0.40 ± 0.02, 0.63 ± 0.03, 0.13 ± 0.02, and 0.04 ± 0.01; n = 11, 14, 8, 17, 7, 14, 13, 15, 22, 19, 10, and 10 cells, respectively. PCC ≥0.4 (dotted line) represents colocalization. (N) Sucrose (10% to 60%) gradient assay shows that TMEM163 coexists with the EE marker EEA1, the LE marker Rab7, and the DG marker LAMP2. Endo, endogenous; IB, immunoblotting.
TMEM163 is localized to perinuclear DG and LE marker–positive compartments (likely DG precursors) and small vesicular structures (likely EEs) in the periphery, but not to AGs in MEG-01 cells. (A-B) MEG-01 cells were cotransfected with TMEM163-GFP or Cherry-TMEM163 and Cherry-VMAT2 (A) or GFP-VMAT2 (B) for 48 hours, respectively. (C) MEG-01 cells were transfected with Cherry-TMEM163 for 48 hours and then treated with 50 μM mepacrine for 30 minutes at 37°C. (D) MEG-01 cells were transfected with TMEM163-GFP for 48 hours and then fixed and stained with antibodies against CD63. (E-F) MEG-01 cells were cotransfected with Cherry-TMEM163 and GFP-Rab7 (E) or GFP-Lamp1 (F) for 48 hours, respectively. (G) MEG-01 cells were cotransfected with GFP-TMEM163 and Cherry-TPCN2 for 48 hours, respectively. (H) MEG-01 cells were transfected with Cherry-TMEM163 for 48 hours and treated with LysoSensor Green DND-189 for 30 minutes at 37°C. (I) MEG-01 cells were transfected with TMEM163-GFP for 48 hours and then fixed and stained with antibodies against EEA1. (J) MEG-01 cells were cotransfected with Cherry-TMEM163 and GFP-Rab5 for 48 hours. (K-L) MEG-01 cells were transfected with GFP-TMEM163 for 48 hours and then fixed and stained with antibodies against P-selectin (K) or VWF (L). Pictures shown in panels A-L are representative confocal images. Outlines of cells are indicated by white lines. Insets show 5× magnified images of the boxed region. Scale bars, 10 μm. (M) Colocalization analysis of results shown in panels A-L; PCC, 0.74 ± 0.04, 0.76 ± 0.02, 0.68 ± 0.04, 0.73 ± 0.03, 0.72 ± 0.03, 0.75 ± 0.02, 0.59 ± 0.04, 0.82 ± 0.02, 0.40 ± 0.02, 0.63 ± 0.03, 0.13 ± 0.02, and 0.04 ± 0.01; n = 11, 14, 8, 17, 7, 14, 13, 15, 22, 19, 10, and 10 cells, respectively. PCC ≥0.4 (dotted line) represents colocalization. (N) Sucrose (10% to 60%) gradient assay shows that TMEM163 coexists with the EE marker EEA1, the LE marker Rab7, and the DG marker LAMP2. Endo, endogenous; IB, immunoblotting.
TMEM163 interacts with BLOC-1
The decreased level of TMEM163 in BLOC-1 mouse mutants (sdy and pa; Figure 1C-D) and an HPS9 patient (Figure 1M) suggests that BLOC-1 may be involved in the trafficking of TMEM163. To determine whether TMEM163 is a cargo of BLOC-1, we tested for possible physical interactions between TMEM163 and the BLOC-1 subunits dysbindin, pallidin, and BLOS1. Coimmunoprecipitation (co-IP) of endogenous proteins showed that dysbindin interacted with TMEM163 (Figure 3A-B). Consistently, due to the deficiencies of dysbindin or TMEM163 in sdy or Tmem163-KO mice platelets, neither the anti-dysbindin nor the anti-TMEM163 antibody precipitated TMEM163 or dysbindin compared with their positive controls (Figure 3C-D). Furthermore, we confirmed that Flag-dysbindin-1A and Flag-BLOS1 (one of the shared subunits of BLOC-1 and BORC) coprecipitated with Myc-TMEM163 by co-IP assays, but not Flag-KXD1 (1 subunit of BORC) (supplemental Figure 4A-D). Moreover, pallidin precipitated with TMEM163 in B6 mice but not in the pa mutant (supplemental Figure 4E). However, the separation of dysbindin in F1 to F8 suggests that dysbindin and TMEM163 do not reside in a tight complex (Figure 2N), implying that TMEM163 may be one of multiple DG proteins transported by BLOC-1. These results suggest that BLOC-1 interacts with TMEM163 during its transport from EEs to perinuclear DG and LE marker–positive compartments (likely DG precursors).
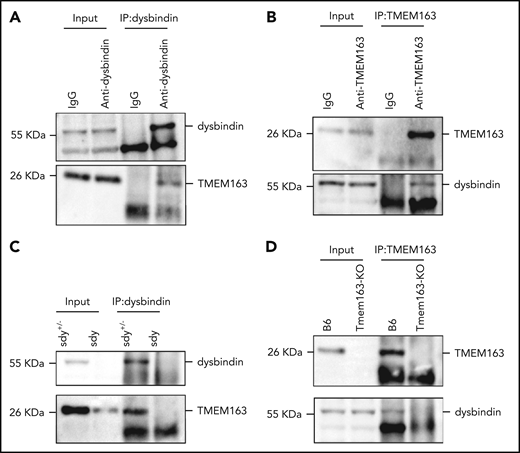
TMEM163 interacts with dysbindin. (A) Co-IP of endogenous platelet proteins show that dysbindin precipitates TMEM163. (B) Co-IP of endogenous platelet proteins show that TMEM163 precipitated dysbindin. The immunoglobulin G (IgG) was used as a negative control. (C) Co-IP of endogenous platelet proteins show that dysbindin precipitated TMEM163 in sdy+/− platelets, but not in sdy platelets. (D) Co-IP of endogenous platelet proteins show that TMEM163 precipitated dysbindin in WT (B6, C57BL/6J) platelets, but not in Tmem163-KO platelets. All the inputs corresponded to 8% of whole platelet lysates.
TMEM163 interacts with dysbindin. (A) Co-IP of endogenous platelet proteins show that dysbindin precipitates TMEM163. (B) Co-IP of endogenous platelet proteins show that TMEM163 precipitated dysbindin. The immunoglobulin G (IgG) was used as a negative control. (C) Co-IP of endogenous platelet proteins show that dysbindin precipitated TMEM163 in sdy+/− platelets, but not in sdy platelets. (D) Co-IP of endogenous platelet proteins show that TMEM163 precipitated dysbindin in WT (B6, C57BL/6J) platelets, but not in Tmem163-KO platelets. All the inputs corresponded to 8% of whole platelet lysates.
TMEM163 deficiency affects the biogenesis of platelet DGs
We further analyzed the morphological characteristics of DTNBP1- and TMEM163-deficient cell lines (DTNBP1-KO [D1, D12, and D24] and TMEM163-KO [T20 and T87] stable cell lines constructed by the CRISPR-Cas9 system, as shown in supplemental Figure 5) by thin-section transmission electron microscopy (TEM). A large number of organelles were present in WT MEG-01 cells after induction with 100 nM TPA for 4 days, including round membranous organelles resembling EEs with less intraluminal contents, LEs with more intraluminal contents and MVBs with intraluminal vesicles. Numerous enlarged round membranous organelles resembling EEs with less intraluminal content increased significantly in DTNBP1-KO (D1 and D24) and TMEM163-KO (T20 and T87) MEG-01 cells (Figure 4A-C). These results suggest that deletion of dysbindin or TMEM163 resulted in the accumulation of large vacuoles.
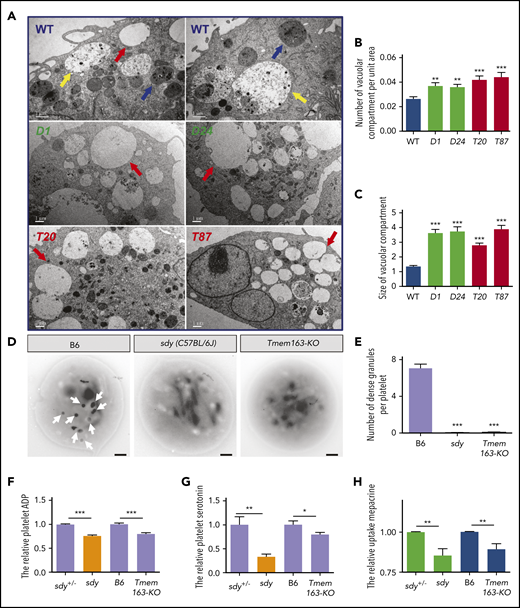
Ultrastructures of DGs in MEG-01 cells and platelets with deficiencies in dysbindin or TMEM163. (A) Thin-section TEM pictures of DTNBP1-KO (D1 and D24) and TMEM163-KO (T20 and T87) MEG-01 cells after induction with 100 nM TPA for 4 days. Red arrows represent round membranous organelles resembling EEs with reduced intraluminal contents, yellow arrows represent round membranous organelles resembling LEs with increased intraluminal contents, and blue arrows represent MVB circular membranous organelles. Scale bars, 1 µm. (B) The average number of vacuolar compartment (red arrows shown in panel A) per unit area (µm2) in DTNBP1-KO (D1 and D24) and TMEM163-KO (T20 and T87) MEG-01 cells are significantly higher than that in control (WT) cells (WT, 0.027 ± 0.002, n = 47; D1, 0.037 ± 0.003, n = 48; D24, 0.036 ± 0.002, n = 52; T20, 0.042 ± 0.003, n = 55; T87, 0.044 ± 0.004, n = 40). (C) The average size of the vacuolar compartment (µm2) in DTNBP1-KO (D1 and D24) and TMEM163-KO (T20 and T87) MEG-01 cells is significantly larger than that in WT cells (WT, 1.343 ± 0.080, n = 158; D1, 3.637 ± 0.234, n = 299; D24, 3.736 ± 0.302, n = 273; T20, 2.791 ± 0.145, n = 382; T87, 3.890 ± 0.245, n = 304). (D-E) TEM pictures of whole-mount platelets of the B6 (WT, C57BL/6J), sdy mutant, and Tmem163-KO mice. Arrows show platelet DGs. Lighter-intensity dots or smaller dots (diameter <100 nm) are not counted as DGs. Scale bars, 0.5 μm. DGs are absent in sdy (0.029 ± 0.020, n = 69) and Tmem163-KO (0.095 ± 0.020, n = 222) mice compared with B6 (7.069 ± 0.451, n = 58). (F-G) Washed platelets were lysed by 100 µL ice-cold lysis buffer, and the lysates used for further enzyme-linked immunosorbent assay were analyzed spectrophotometrically at 450 nm. The content of platelet ADP (sdy vs Tmem163-KO, 75.71% vs 79.77%, n = 4, 13, respectively) (F) and serotonin (sdy vs Tmem163-KO, 33.01% vs 79.51%, n = 6, 10, respectively) (G) of sdy, Tmem163-KO mice were calculated and plotted relative to their control littermates (sdy+/− or B6). (H) Platelets were incubated with 1.7 μM mepacrine at 37°C for 30 minutes and then analyzed by flow cytometry. The mepacrine uptake (green) fluorescence of sdy and Tmem163-KO was expressed as the percentage (mean ± SEM) of the mean normalized value for control mice (sdy vs Tmeme163-KO, 85.41% vs 89.29%, n = 9, 8, respectively). *P < .05; **P < .01; ***P < .001.
Ultrastructures of DGs in MEG-01 cells and platelets with deficiencies in dysbindin or TMEM163. (A) Thin-section TEM pictures of DTNBP1-KO (D1 and D24) and TMEM163-KO (T20 and T87) MEG-01 cells after induction with 100 nM TPA for 4 days. Red arrows represent round membranous organelles resembling EEs with reduced intraluminal contents, yellow arrows represent round membranous organelles resembling LEs with increased intraluminal contents, and blue arrows represent MVB circular membranous organelles. Scale bars, 1 µm. (B) The average number of vacuolar compartment (red arrows shown in panel A) per unit area (µm2) in DTNBP1-KO (D1 and D24) and TMEM163-KO (T20 and T87) MEG-01 cells are significantly higher than that in control (WT) cells (WT, 0.027 ± 0.002, n = 47; D1, 0.037 ± 0.003, n = 48; D24, 0.036 ± 0.002, n = 52; T20, 0.042 ± 0.003, n = 55; T87, 0.044 ± 0.004, n = 40). (C) The average size of the vacuolar compartment (µm2) in DTNBP1-KO (D1 and D24) and TMEM163-KO (T20 and T87) MEG-01 cells is significantly larger than that in WT cells (WT, 1.343 ± 0.080, n = 158; D1, 3.637 ± 0.234, n = 299; D24, 3.736 ± 0.302, n = 273; T20, 2.791 ± 0.145, n = 382; T87, 3.890 ± 0.245, n = 304). (D-E) TEM pictures of whole-mount platelets of the B6 (WT, C57BL/6J), sdy mutant, and Tmem163-KO mice. Arrows show platelet DGs. Lighter-intensity dots or smaller dots (diameter <100 nm) are not counted as DGs. Scale bars, 0.5 μm. DGs are absent in sdy (0.029 ± 0.020, n = 69) and Tmem163-KO (0.095 ± 0.020, n = 222) mice compared with B6 (7.069 ± 0.451, n = 58). (F-G) Washed platelets were lysed by 100 µL ice-cold lysis buffer, and the lysates used for further enzyme-linked immunosorbent assay were analyzed spectrophotometrically at 450 nm. The content of platelet ADP (sdy vs Tmem163-KO, 75.71% vs 79.77%, n = 4, 13, respectively) (F) and serotonin (sdy vs Tmem163-KO, 33.01% vs 79.51%, n = 6, 10, respectively) (G) of sdy, Tmem163-KO mice were calculated and plotted relative to their control littermates (sdy+/− or B6). (H) Platelets were incubated with 1.7 μM mepacrine at 37°C for 30 minutes and then analyzed by flow cytometry. The mepacrine uptake (green) fluorescence of sdy and Tmem163-KO was expressed as the percentage (mean ± SEM) of the mean normalized value for control mice (sdy vs Tmeme163-KO, 85.41% vs 89.29%, n = 9, 8, respectively). *P < .05; **P < .01; ***P < .001.
We speculated that BLOC-1 participates in the biogenesis of DGs by regulating the transport of TMEM163 to DGs, and loss-of-function mutants of TMEM163 or BLOC-1 have similar phenotypes. To test this hypothesis, we constructed Tmem163 knockout mice (Tmem163-KO) using the CRISPR-Cas9 system (supplemental Figure 6A-B). Both the tail genomic PCR and western blotting assays proved that the Tmem163 gene was successfully deleted (supplemental Figure 6C-E). Through whole-mount TEM experiments, which has been used as the gold standard method for diagnosing platelet DG deficiencies,59-62 we found that DGs in Tmem163-KO mice were absent, which is similar to sdy (C57BL/6J) mice (Figure 4D-E), suggesting that TMEM163 deficiency likely affects the biogenesis of platelet DGs.
Tmem163-KO (n = 6) and C57BL/6J (B6) (n = 8) mice did not differ significantly in platelet count, mean platelet volume, and other hematological parameters (supplemental Table 2). We observed no abnormalities in the surface expression of CD41, CD42b (GPIbα), and GPVI in sdy and Tmem163-KO platelets compared with their controls (supplemental Figure 7).
DGs concentrate small molecules such as serotonin, adenosine 5′-diphosphate (ADP), and Ca2+.63,64 Consistently, similar to sdy mutant, Tmem163-KO platelets displayed reduced ADP (Figure 4F) and serotonin (Figure 4G) relative to their littermate controls. Mepacrine, a green fluorescent dye, has a high affinity for adenine nucleotides and can be taken up by platelet DGs rapidly and selectively. Platelets of HPS patients have a significantly reduced uptake of mepacrine into DGs.65-67 Similar to platelets of sdy mice, Tmem163-KO mouse platelets displayed significantly deficient uptake of mepacrine relative to platelets of littermate controls with a high dose of mepacrine (1.7 µM) (Figure 4H). In addition, we noticed that the defects in serotonin storage and mepacrine uptake were less profound in Tmem163-KO platelets than in sdy platelets, suggesting that TMEM163 plays a more specific role in DG biogenesis than BLOC-1 does.
Furthermore, we analyzed the ultrastructure of organelles in platelets of Tmem163-KO, sdy and pa (another BLOC-1 mutant68 ) mice. Based on the density of the dense core of round membranous compartments, the organelles of B6 mouse platelets were divided into 2 types; type I has low electron density (average gray value >50), and type II has a higher electron density (average gray value <50) (supplemental Figure 8F). Statistical analysis of type I and type II compartments in Tmem163-KO, sdy and pa mutant mice, and their corresponding control mice, showed that Tmem163-KO, sdy and pa mutant mice displayed an abnormal type I accumulation and decreased type II compartments (supplemental Figure 8A-E,G-J). We speculate that the type I compartment represents an immature DG and the type II compartment represents a mature DG. This requires further confirmation by future immunogold labeling assays.
Moreover, Tmem163-KO platelets displayed normal steady-state levels of AG content and membrane proteins (basic fibroblast growth factor, endostatin, PF4, P-selectin, thrombospondin, and VWF) (supplemental Figure 9) and showed a significantly weaker endocytosis of fibrinogen (supplemental Figure 10), indicating that TMEM163 deficiency does not apparently affect the biogenesis of platelet AGs. Taken together, these results demonstrate that TMEM163 deficiency affects the biogenesis of platelet DGs, but not AGs. This finding is similar to that observed in BLOC-1 deficiency.
TMEM163 deficiency impairs platelet DG secretion
We next investigated possible roles of TMEM163 in the secretion of DGs by detecting adenosine triphosphate (ATP) release and in the secretion of AGs by measuring CD62p exposure and PF4 release. At various concentrations of collagen, sdy mutant and Tmem163-KO platelets both displayed significantly defective release of ATP compared with their littermate controls (sdy+/− or B6 mice) (Figure 5A; supplemental Table 3). Consistently, at various concentrations of thrombin (0.25, 1 U/mL), sdy mutant and Tmem163-KO platelets displayed significantly defective release of ATP (Figure 5B; supplemental Table 3). Both sdy and Tmem163-KO platelets showed impaired ATP release relative to controls in response to both collagen and thrombin. However, the defects in Tmem163-KO mice were milder than those in sdy mice, which is consistent with the milder serotonin storage and mepacrine uptake defects in the former, supporting that TMEM163 is one of multiple DG proteins transported by BLOC-1.
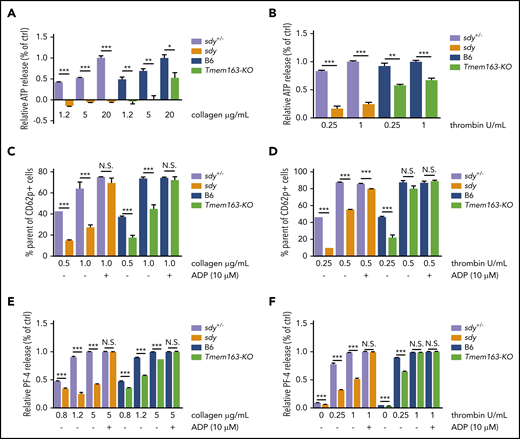
Sdy and Tmem163-KO platelets display significant impairment of DG release and AG secretion. (A-B) Washed platelets from sdy+/−, sdy, B6 (WT, C57BL/6J) or Tmem163-KO mice were stimulated with various concentrations of collagen (1.2, 5, or 20 μg/mL) (A) or thrombin (0.25 or 1 U/mL) (B) and secretion of ATP assessed by ATP determination kit. Data represent the relative ATP release as the percentage (mean ± SEM) of the mean normalized value in maximal dose stimulation for their control mice. (C-D) Washed platelets from sdy+/−, sdy, B6, or Tmem163-KO mice were labeled with fluorescein isothiocyanate-CD62p antibody and stimulated with various concentrations of collagen (0.5 or 1.0 μg/mL, sdy vs Tmem163-KO, n = 6, 6, respectively) (C) or thrombin (0.25 or 0.5 U/mL, sdy vs Tmem163-KO, n = 3, 6, respectively) (D) for 5 minutes in the absence or presence of 10 μM ADP and then measured by flow cytometry for CD62p surface expression. Data represent the percentage of platelets with labeling above the background level observed on unstimulated platelets. (E-F) Washed platelets from sdy+/−, sdy, B6, or Tmem163-KO mice were stimulated with various concentrations of collagen (0.8, 1.2, or 5.0 μg/mL) (E) or thrombin (0, 0.25, or 1 U/mL) (F) for 5 minutes in the absence or presence of 10 μM ADP, and then supernatants were collected and measured by enzyme-linked immunosorbent assay for PF-4 (sdy vs Tmem163-KO, n = 4, 4, respectively.). Data shown represent the relative PF4 release as the percentage (mean ± SEM) of the mean normalized value in maximal dose stimulation for their control mice. *P < .05; **P < .01; ***P < .001.
Sdy and Tmem163-KO platelets display significant impairment of DG release and AG secretion. (A-B) Washed platelets from sdy+/−, sdy, B6 (WT, C57BL/6J) or Tmem163-KO mice were stimulated with various concentrations of collagen (1.2, 5, or 20 μg/mL) (A) or thrombin (0.25 or 1 U/mL) (B) and secretion of ATP assessed by ATP determination kit. Data represent the relative ATP release as the percentage (mean ± SEM) of the mean normalized value in maximal dose stimulation for their control mice. (C-D) Washed platelets from sdy+/−, sdy, B6, or Tmem163-KO mice were labeled with fluorescein isothiocyanate-CD62p antibody and stimulated with various concentrations of collagen (0.5 or 1.0 μg/mL, sdy vs Tmem163-KO, n = 6, 6, respectively) (C) or thrombin (0.25 or 0.5 U/mL, sdy vs Tmem163-KO, n = 3, 6, respectively) (D) for 5 minutes in the absence or presence of 10 μM ADP and then measured by flow cytometry for CD62p surface expression. Data represent the percentage of platelets with labeling above the background level observed on unstimulated platelets. (E-F) Washed platelets from sdy+/−, sdy, B6, or Tmem163-KO mice were stimulated with various concentrations of collagen (0.8, 1.2, or 5.0 μg/mL) (E) or thrombin (0, 0.25, or 1 U/mL) (F) for 5 minutes in the absence or presence of 10 μM ADP, and then supernatants were collected and measured by enzyme-linked immunosorbent assay for PF-4 (sdy vs Tmem163-KO, n = 4, 4, respectively.). Data shown represent the relative PF4 release as the percentage (mean ± SEM) of the mean normalized value in maximal dose stimulation for their control mice. *P < .05; **P < .01; ***P < .001.
HPS6 deficiency displays impaired protein disulfide isomerase secretion and impaired exocytosis of AGs69 and affects the tubulation and secretion of VWF.70 Platelets from the HPS mutants pearl (pe), pallid (pa), and light ear (le) display impairment of AG secretion, which is regarded as secondary to deficient DG secretion.71 Likewise, flow cytometry analysis demonstrated that sdy mutant and Tmem163-KO platelets displayed significant impairment of CD62p exposure at low doses of collagen (0.5 or 1.0 μg/mL) or thrombin (0.25 U/mL) compared with their littermate controls. Notably, the defects in Tmem163-KO mice were relatively milder than those in sdy mice. Impaired AG secretion could be rescued by high doses of agonists or exogenous ADP when there is no primary AG deficiency (Figure 5C-D; supplemental Figures 11 and 12). At higher doses of thrombin (0.5 U/mL) or with ADP supplement, CD62p levels revealed no significant differences (Figure 5C-D). Moreover, at various concentrations of collagen (0.8, 1.2, or 5 μg/mL) or at low doses of thrombin (0.25 U/mL), sdy mutant and Tmem163-KO platelets displayed significantly defective PF4 release. However, with a higher dose of thrombin (1 U/mL) or ADP supplement, there was no significant difference in PF4 release (Figure 5E-F). Taken together, both sdy and Tmem163-KO platelets showed impaired AG secretion relative to controls in response to both collagen and thrombin, suggesting that the impairment of AG secretion in sdy and Tmem163-KO platelets is secondary to deficient DG secretion and that there are no bona fide AG defects in both mutants.
TMEM163 deficiency impairs platelet function
To determine whether the defects of DG secretion resulted in abnormal platelet aggregation, we evaluated the aggregation of washed platelets from sdy and Tmem163-KO mice stimulated with various doses of agonists in a turbidimetric aggregometer. At different doses of collagen (1.2 or 2.4 μg/mL), sdy and Tmem163-KO mice showed significant impairment of platelet aggregation compared with their littermate controls (Figure 6A-B,E). Consistently, at different doses of thrombin (0.025 or 0.05 U/mL), sdy and Tmem163-KO mice showed significant impairment of platelet aggregation (Figure 6C-D,F). We further tested hemostasis in Tmem163-KO mice by recording tail-bleeding times. Tmem163-KO mice had significant prolongation in bleeding time (Figure 6G), suggesting that TMEM163 deficiency results in defective platelet aggregation and prolonged bleeding resembling the sdy mutant.
![Sdy and Tmem163-KO display significant impairment of platelet aggregation and prolonged bleeding time. (A-F) Washed platelets from sdy+/−, sdy, B6 (WT, C57BL/6J), or Tmem163-KO mice were stimulated with various concentrations of collagen (sdy+/− vs sdy, 39.38% vs 11.04% [1.2 μg/mL], and 71.05% vs 9.71% [2.4 μg/mL], n = 4, 4, 3, and 3, respectively; B6 vs Tmem163-KO, 51.50% vs 13.80% [1.2 μg/mL], and 80.42% vs 33.65% [2.4 μg/mL], n = 4, 4, 4, and 4, respectively) (A-B,E) or thrombin (sdy+/− vs sdy, 31.91% vs 6.53% [0.025 U/mL], and 68.03% vs 27.78% [0.05 U/mL], n = 4, 5, 3, and 3, respectively; B6 vs Tmem163-KO, 21.60% vs 0.99% [0.025 U/mL], 65.01% vs 25.44% [0.05 U/mL], n = 9, 7, 7, and 6, respectively) (C-D,F) and aggregation assessed by a turbidimetric aggregometer. (G) Tail-bleeding assays showed Tmem163-KO mice have prolonged bleeding times compared with B6 mice (B6, 135 ± 27.59 s; n = 9; Tmem163-KO, >900 s; n = 12). Student t test, *P < .05; **P < .01; ***P < .001. OD, optic density.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/13/10.1182_blood.2020007389/1/m_bloodbld2020007389f6.png?Expires=1770174213&Signature=omIUwHuK2TCZkfjur7LFY4S-tq0Yy8caNbWf-lOj2eXa97bj~Ax6ZN5QiAAw5OrefbCVuhoWxi7vhlIemqW29psUp8DOZlKbgcGzBVW2eC9t4WLx1JBgVuNSMsepGDy7LcZ2TM60whjLRVPfzEIPMKz~6oRetmf7398Mwa5fdryR5pD0uD-3eL8X3gxYvzcYI4VTy-uNO3fFNfRJpyfR0VgbpzRKF2ndrmNM66SM0X6DC0x4GE8~i-ZaVzKQElLrJsVEgYodAQtZmnaq7fjIhjkGBvhuVciZEiSyxDQGBXRFUEfRNJDLk1CnK45XeAUCcfQzkRewepmFrbd57oTEag__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Sdy and Tmem163-KO display significant impairment of platelet aggregation and prolonged bleeding time. (A-F) Washed platelets from sdy+/−, sdy, B6 (WT, C57BL/6J), or Tmem163-KO mice were stimulated with various concentrations of collagen (sdy+/− vs sdy, 39.38% vs 11.04% [1.2 μg/mL], and 71.05% vs 9.71% [2.4 μg/mL], n = 4, 4, 3, and 3, respectively; B6 vs Tmem163-KO, 51.50% vs 13.80% [1.2 μg/mL], and 80.42% vs 33.65% [2.4 μg/mL], n = 4, 4, 4, and 4, respectively) (A-B,E) or thrombin (sdy+/− vs sdy, 31.91% vs 6.53% [0.025 U/mL], and 68.03% vs 27.78% [0.05 U/mL], n = 4, 5, 3, and 3, respectively; B6 vs Tmem163-KO, 21.60% vs 0.99% [0.025 U/mL], 65.01% vs 25.44% [0.05 U/mL], n = 9, 7, 7, and 6, respectively) (C-D,F) and aggregation assessed by a turbidimetric aggregometer. (G) Tail-bleeding assays showed Tmem163-KO mice have prolonged bleeding times compared with B6 mice (B6, 135 ± 27.59 s; n = 9; Tmem163-KO, >900 s; n = 12). Student t test, *P < .05; **P < .01; ***P < .001. OD, optic density.
Sdy and Tmem163-KO display significant impairment of platelet aggregation and prolonged bleeding time. (A-F) Washed platelets from sdy+/−, sdy, B6 (WT, C57BL/6J), or Tmem163-KO mice were stimulated with various concentrations of collagen (sdy+/− vs sdy, 39.38% vs 11.04% [1.2 μg/mL], and 71.05% vs 9.71% [2.4 μg/mL], n = 4, 4, 3, and 3, respectively; B6 vs Tmem163-KO, 51.50% vs 13.80% [1.2 μg/mL], and 80.42% vs 33.65% [2.4 μg/mL], n = 4, 4, 4, and 4, respectively) (A-B,E) or thrombin (sdy+/− vs sdy, 31.91% vs 6.53% [0.025 U/mL], and 68.03% vs 27.78% [0.05 U/mL], n = 4, 5, 3, and 3, respectively; B6 vs Tmem163-KO, 21.60% vs 0.99% [0.025 U/mL], 65.01% vs 25.44% [0.05 U/mL], n = 9, 7, 7, and 6, respectively) (C-D,F) and aggregation assessed by a turbidimetric aggregometer. (G) Tail-bleeding assays showed Tmem163-KO mice have prolonged bleeding times compared with B6 mice (B6, 135 ± 27.59 s; n = 9; Tmem163-KO, >900 s; n = 12). Student t test, *P < .05; **P < .01; ***P < .001. OD, optic density.
Tmem163-KO mice and HPS mouse mutants of BLOC-1, BLOC-2, and AP-3 show defective Zn2+ homeostasis
To further define the role of TMEM163 in the biogenesis of DGs, we investigated its function in megakaryocytes. TMEM163 has been shown to bind Zn2+ presumably as a proton-dependent Zn2+ transporter.45 During blood clotting, activated platelets release a large amount of stored Zn2+ to the bloodstream.31,72,73 Once platelets are stimulated, the 2 types of secretory granules (AGs and DGs) activate. AGs store protein-bound Zn2+,74,75 but it is uncertain whether Zn2+ ions are present in DGs. When MEG-01 cells expressing Cherry-VMAT2 were treated with the Zn2+ indicator FluoZin-3, we found VMAT2-positive organelles enriched with FluoZin-3 dye (Figure 7A). Furthermore, we found that TMEM163 may act as a limiting membrane protein that packages FluoZin-3 dye (Figure 7B-C). These results suggest that perinuclear DG and LE marker–positive compartments (likely DG precursors) store free Zn2+.
![Defective Zn2+homeostasis in platelets from Tmem163-KO mice and HPS mouse models of BLOC-1, BLOC-2 and AP-3. (A-B) MEG-01 cells were transfected with Cherry-VMAT2 (A) or Cherry-TMEM163 (B) for 48 hours then incubated with 2 μM Zn2+ indicator (FluoZin-3) for 30 minutes at 37°C. Pictures are representative confocal images. Outline of the cell is indicated by the white line. Insets show 5× magnified images of the boxed region. Scale bars, 10 μm. (C) MEG-01 cells were transfected with Cherry-TMEM163 for 48 hours and then incubated with 2 μM Zn2+ indicator (FluoZin-3) for 30 minutes at 37°C. Picture (left) is representative maximum intensity projection image in X-Y, X-Z, and Y-Z planes. Pictures (right) are Z-stack images in different dimensions. The arrows represent the classical colocalization of Cherrry-TMEM163 with FluoZin-3. (D-I) Washed mutant and control mice platelets were incubated with 2.5 µM FluoZin-3 and stimulated with 0.01 U/mL thrombin, and the kinetic curve of fluorescence changes was recorded by flow cytometry (appending the first 50 seconds [resting stage] to 400 seconds [activated stage]). Data shown are the quantitative results (D) of normalized FluoZin3 mean fluorescence intensity relative to each control at resting conditions and representative kinetic curves of Zn2+ release after thrombin treatment (E-I). B6 vs Tmem163-KO, n = 11, 11; sdy+/− vs sdy, n = 10, 10; pa+/− vs pa, n = 12; 12, ru+/− vs ru, n = 12, 12; and pe+/− vs pe, n = 12, 12, respectively. MFI, mean fluorescence intensity. Mean ± SEM, **P < .01; ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/13/10.1182_blood.2020007389/1/m_bloodbld2020007389f7.png?Expires=1770174213&Signature=Huvi9Ox8FCCpdmMm1tqFMM63n9etsq1un4TTPxEbaqHIKpxf5l1b7vDlB5BtsHhZDUy2Jg53lxOO12gAghJp7Z1WZwfEAdPy49u0oiK1JD0oAe3AT33B63SxI0eCx~wmxuqn-X~EqwY3Rgz0ua2PlqmkFkdo1~HnhoG6uo~d-3APmQd4PIjGKLDJa1hKxR8EcV-Mmrku0mb-jNwl5b6rwV~JcufYORhryIaqMMCyPQhQ1E0amUY-TXChFMnCS8MJlfyURrbmA2PZoU6ViXwXuOBj0XIsOArKiYWdfMGtvxQ64SNbDXzM5ikRnE7BEScyAjDSwVwXAa6gtFHkdirWTw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Defective Zn2+homeostasis in platelets from Tmem163-KO mice and HPS mouse models of BLOC-1, BLOC-2 and AP-3. (A-B) MEG-01 cells were transfected with Cherry-VMAT2 (A) or Cherry-TMEM163 (B) for 48 hours then incubated with 2 μM Zn2+ indicator (FluoZin-3) for 30 minutes at 37°C. Pictures are representative confocal images. Outline of the cell is indicated by the white line. Insets show 5× magnified images of the boxed region. Scale bars, 10 μm. (C) MEG-01 cells were transfected with Cherry-TMEM163 for 48 hours and then incubated with 2 μM Zn2+ indicator (FluoZin-3) for 30 minutes at 37°C. Picture (left) is representative maximum intensity projection image in X-Y, X-Z, and Y-Z planes. Pictures (right) are Z-stack images in different dimensions. The arrows represent the classical colocalization of Cherrry-TMEM163 with FluoZin-3. (D-I) Washed mutant and control mice platelets were incubated with 2.5 µM FluoZin-3 and stimulated with 0.01 U/mL thrombin, and the kinetic curve of fluorescence changes was recorded by flow cytometry (appending the first 50 seconds [resting stage] to 400 seconds [activated stage]). Data shown are the quantitative results (D) of normalized FluoZin3 mean fluorescence intensity relative to each control at resting conditions and representative kinetic curves of Zn2+ release after thrombin treatment (E-I). B6 vs Tmem163-KO, n = 11, 11; sdy+/− vs sdy, n = 10, 10; pa+/− vs pa, n = 12; 12, ru+/− vs ru, n = 12, 12; and pe+/− vs pe, n = 12, 12, respectively. MFI, mean fluorescence intensity. Mean ± SEM, **P < .01; ***P < .001.
Defective Zn2+homeostasis in platelets from Tmem163-KO mice and HPS mouse models of BLOC-1, BLOC-2 and AP-3. (A-B) MEG-01 cells were transfected with Cherry-VMAT2 (A) or Cherry-TMEM163 (B) for 48 hours then incubated with 2 μM Zn2+ indicator (FluoZin-3) for 30 minutes at 37°C. Pictures are representative confocal images. Outline of the cell is indicated by the white line. Insets show 5× magnified images of the boxed region. Scale bars, 10 μm. (C) MEG-01 cells were transfected with Cherry-TMEM163 for 48 hours and then incubated with 2 μM Zn2+ indicator (FluoZin-3) for 30 minutes at 37°C. Picture (left) is representative maximum intensity projection image in X-Y, X-Z, and Y-Z planes. Pictures (right) are Z-stack images in different dimensions. The arrows represent the classical colocalization of Cherrry-TMEM163 with FluoZin-3. (D-I) Washed mutant and control mice platelets were incubated with 2.5 µM FluoZin-3 and stimulated with 0.01 U/mL thrombin, and the kinetic curve of fluorescence changes was recorded by flow cytometry (appending the first 50 seconds [resting stage] to 400 seconds [activated stage]). Data shown are the quantitative results (D) of normalized FluoZin3 mean fluorescence intensity relative to each control at resting conditions and representative kinetic curves of Zn2+ release after thrombin treatment (E-I). B6 vs Tmem163-KO, n = 11, 11; sdy+/− vs sdy, n = 10, 10; pa+/− vs pa, n = 12; 12, ru+/− vs ru, n = 12, 12; and pe+/− vs pe, n = 12, 12, respectively. MFI, mean fluorescence intensity. Mean ± SEM, **P < .01; ***P < .001.
Next, we incubated DTNBP1-KO and TMEM163-KO cell lines with FluoZin-3 in the absence or presence of 100 μM ZnCl2. We observed higher intracellular Zn2+ accumulation in granule-like structures of DTNBP1-KO (D1, D24) and TMEM163-KO (T20, T87) MEG-01 cells (supplemental Figure 13). In addition, washed platelets from sdy and Tmem163-KO mice also showed higher Zn2+ accumulation compared with platelets of their littermate controls under resting conditions. Similarly, the resting platelets of other HPS mouse models of BLOC-1 (pa), BLOC-2 (ru), and AP-3 (pe) mice showed higher Zn2+ accumulation (Figure 7D), consistent with the decreased TMEM163 expression.
Once platelets are activated by thrombin, Zn2+ is rapidly released from granules. Our data displayed similar kinetics of decrease in platelet Zn2+ levels in all mutant and control mice after activation by thrombin (Figure 7E-H), suggesting that Zn2+ is mostly stored in thrombin-triggered releasable granules. However, Zn2+ in AP-3 (pe)–deficient mice did not decline to basal levels (Figure 7I). One explanation is that the amount of thrombin used in this assay was not sufficient to release the highly accumulated Zn2+ in pe mice, as the fold change in pe platelets was the highest among these mutant mice (Figure 7D). Zn2+ accumulation in the platelets of these mutant mice may result from the defects in other Zn2+ transporters (ZnT/ZIP family members) residing in the organelles of platelets. However, our quantitative reverse transcription PCR results showed normal messenger RNA expression of ZnT (ZNT1, ZNT5, ZNT6, ZNT7, and ZNT9) and ZIP (ZIP1, ZIP4, ZIP6, ZIP7, ZIP9, and ZIP10) family members in mature megakaryocytes from Tmem163-KO mice relative to B6 mice (supplemental Figure 14). This suggests that TMEM163 deficiency may be the major contributor of Zn2+ accumulation in megakaryocytes and platelets.
Next, to explore whether decreasing Zn2+ accumulation in sdy and Tmem163-KO platelets would restore normal platelet aggregation, we incubated washed platelets with FluoZin-3 in the presence of a Zn2+ chelator, TPEN, and recorded the kinetics of aggregation during resting and activated states. Both resting and activated platelets of sdy and Tmem163-KO displayed reduced FluoZin-3 intensities with the addition of TPEN (supplemental Figure 15A-C), suggesting that TPEN effectively removes Zn2+ from platelets. We then incubated sdy and Tmem163-KO platelets with either 20 μM or 50 μM TPEN (dimethyl sulfoxide as a negative control). After stimulation with 0.05 U/mL thrombin, we found that TPEN inhibited platelet aggregation in a dose-dependent manner and did not restore the impairment of platelet aggregation in sdy and Tmem163-KO mice (supplemental Figure 15D-G), suggesting that the mutant defects occur in DG biogenesis, but not in DG secretion.
Taken together, these results demonstrate that deficiency of DTNBP1 or TMEM163 compromises intracellular organellar Zn2+ homeostasis in platelets and that defective Zn2+ homeostasis is highly correlated with DG biogenesis.
Discussion
Using different HPS mouse models and HPS patients, we identified a previously unreported DG Zn2+ transporter, TMEM163, which is significantly reduced in BLOC-1 (sdy and pa), BLOC-2 (ru), or AP-3 (pe) deficient mice and HPS patients (HPS2, HPS3, HPS5, HPS6, or HPS9). Furthermore, we determined that BLOC-1 interacts with TMEM163 during its transport from EEs to perinuclear DG and LE marker–positive compartments (likely DG precursors). Therefore, all of these mutant mice have defects in DG biogenesis and secretion, resulting in prolonged bleeding times and impaired platelet aggregation. Finally, similarly to the deficiency of BLOC-1, BLOC-2, or AP-3, which results in abnormal platelet intracellular organellar Zn2+ homeostasis of platelets, TMEM163 deficiency alters platelet Zn2+ homeostasis. These new findings are important for understanding the LRO defects in HPS and platelet storage pool deficiency.
From studies on melanosomes, it is reasonable to propose that HPACs are involved in platelet DG biogenesis by regulating the trafficking of multiple DG cargoes. In this study, we identified a novel membrane protein (likely a DG membrane protein), TMEM163, which interacts with BLOC-1 and is one of the multiple DG proteins transported by BLOC-1. Notably, our data demonstrated that TMEM163 was partially localized to EEs and was deficient in BLOC-1, BLOC-2, or AP-3 mutant mice but at normal levels in BLOC-3 or HOPS mutant mice. There are reports that in melanocytes and neurons, BLOC-1 and AP-3 act upstream of BLOC-2 in cargo trafficking, whereas BLOC-3 and HOPS act in different trafficking pathways.1,2,76 Although it has been reported that RAB32 and RAB38 are required for the biogenesis of DGs,10,16 both the expression of Rab32 and Rab38 in the platelets of Tmem163-KO mice showed no significant changes compared with their control mice (supplemental Figure 16). In addition, it has been reported that BLOC-1 physically interacts with BLOC-2 or AP-3 to facilitate TYRP1 trafficking from endosomes to melanosomes.12 Our results suggest that BLOC-1, BLOC-2, and AP-3 are functionally required for TMEM163 transport, similar to some cargoes of melanosomes57,77 and SVs.78,79
Several other DG integral membrane proteins such as LAMP2, CD63, and VMAT210,19 have been identified, but the possible involvement of these proteins in DG biogenesis is unknown. However, the substantial combined evidence of whole-mount TEM, thin-section TEM, and DG contents presented here strongly support the critical involvement of TMEM163 in DG biogenesis. In summary, our data are consistent with a role of HPACs in the biogenesis of LROs, in which BLOC-1, BLOC-2, and AP-3 function in trafficking of TMEM163 from endosomes to perinuclear DG and LE marker–positive compartments (likely DG precursors). Considering that TMEM163 is specifically deficient in BLOC-1, BLOC-2, or AP-3 mouse models and HPS patients, we propose that TMEM163 or platelet Zn2+ concentration could be a new marker for the diagnosis and genotyping of HPS subtypes.
Platelet whole-mount TEM has long been the gold standard method for diagnosing platelet DG deficiencies.59-62 As expected, Tmem163-KO platelets showed the typical “DG null” phenotype by this procedure. Interestingly, through thin-section TEM of platelets, we found that a deficiency of TMEM163, dysbindin, or pallidin causes the accumulation of the type I compartment (likely immature DGs) and a decrease in the type II compartment (likely mature DGs), which requires further future confirmation by immunogold labeling assays. Further analysis by thin-section TEM showed both DTNBP1-KO and TMEM163-KO MEG-01 cell lines have increased enlarged vacuoles, which may represent immature DGs or DG precursors in megakaryocytes. However, the underlying mechanisms of DG maturation during megakaryocyte differentiation require further investigation.
A previous report demonstrated that TMEM163 lies within the limiting membrane of SVs.41 Both SVs and DGs belong to LROs.2 It has been reported that TRPML1 interacts with TMEM163 to regulate the trafficking of TMEM163 from the plasma membrane to endocytic compartments, lysosomes, or SVs. TMEM163 was proposed as a zinc (Zn2+)/proton (H+) exchanger and as a cooperative ionic channel with TRPML1 in lysosomes. TMEM163 functions in influxing Zn2+ into the cytoplasm from the extracellular matrix and the lumen of vesicular compartments,43,44 indicating that TMEM163 acts as a ZIP protein and not as a ZNT protein. However, another study reported that TMEM163 effluxes Zn2+ from the cytosol. Thus, TMEM163 was designated as ZNT11, a new member of the ZNT family.48 When SV31 is reconstituted in liposomes, TMEM163 assembles into a dimer and functions as a proton-dependent Zn2+ transporter, similar to ZNT3.45 Our investigation indicated that deficiency of TMEM163 results in abnormally high intracellular Zn2+ content (accumulation in granule-like structures), thus affecting the biogenesis and function of DGs. We speculate that TMEM163 acts as a ZIP protein on DGs as Zn2+ is accumulated in granule-like structures when TMEM163 is deficient (Figure 7). Accumulated Zn2+ in platelets with a TMEM163 deficiency (or BLOC-1, BLOC-2, or AP-3 deficiency) may disrupt the ion homeostasis that is essential for DG biogenesis and secretion.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Richard T. Swank, who provided the mutant mice in this study and proofread the manuscript; Runlin Z. Ma, who provided the pSpCas9 (BB)-2A-GFP (PX458) vector; Fangyuan Nie, who helped to analyze quantitative reverse transcription PCR data; and Chang Zhang, who provided the pEGFP-C2-Rab5, pEGFP-C2-Rab7, and pEGFP-C2-Lamp1 vectors. The authors thank Jiaying Yu, Jiran Lu, and Huipeng Wang for their help breeding several mutant mice.
This work was supported by grants from the Ministry of Science and Technology of China (2019YFA0802104) and the National Natural Science Foundation of China (31800977, 91954000, 31830054, 91539204, and 31900496).
Authorship
Contribution: Y.Y. and W.L. designed the research, analyzed the data and wrote the manuscript; Y.Y. performed most of the experiments; T. Liu and A.W. recruited all the subjects and collected the written informed consents; Y.C. participated in Tmem163-KO mouse generation; W.Z. and Q.C. assisted in the design of platelet aggregation assays; T. Li provided technical support for flow cytometry; L.Y. provided technical support for electron microscopy; and X.H. and Y.W. helped analyze mass spectrometry data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wei Li, Beijing Children’s Hospital, Capital Medical University, 56 Nan Li Shi Rd, Xicheng District, Beijing 100045, China; e-mail: liwei@bch.com.cn.
