

Key Points
A missense mutation in the DNMT1 BAH1 domain epigenetically derepresses γ-globin expression and ameliorates β-thalassemia severity.
Missense DNMT1 variant S878F is identified as a novel genetic modifier implicated in high HbF production and, thus, the mild phenotype.

Abstract
DNA methyltransferase 1 (DNMT1) is a major epigenetic regulator of the formation of large macromolecular complexes that repress human γ-globin expression by maintaining DNA methylation. However, very little is known about the association of DNMT1 variants with β-thalassemia phenotypes. We systematically investigated associations between variants in DNMT1 and phenotypes in 1142 β-thalassemia subjects and identified a novel missense mutation (c.2633G>A, S878F) in the DNMT1 bromo-adjacent homology-1 (BAH1) domain. We functionally characterized this mutation in CD34+ cells from patients and engineered HuDEP-2 mutant cells. Our results demonstrate that DNMT1 phosphorylation is abrogated by substituting serine with phenylalanine at position 878, resulting in lower stability and catalytic activity loss. S878F mutation also attenuated DNMT1 interactions with BCL11A, GATA1, and HDAC1/2, and reduced recruitment of DNMT1 to the γ-globin (HBG) promoters, leading to epigenetic derepression of γ-globin expression. By analyzing the F-cell pattern, we demonstrated that the effect of DNMT1 mutation on increased fetal hemoglobin (HbF) is heterocellular. Furthermore, introduction of S878F mutation into erythroid cells by clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) recapitulated γ-globin reactivation. Thus, the natural S878F DNMT1 mutation is a novel modulator of HbF synthesis and represents a potential new therapeutic target for β-hemoglobinopathies.
Introduction
For greater insight into the mechanism of hematopoietic diseases, finding genetic modifiers that account for the clinical variability of β-hemoglobinopathies, including hemoglobin E (HbE)/β-thalassemia, is of crucial importance.1,2 A group of genetic modifiers involved in the regulation of fetal hemoglobin (HbF) has been shown to be responsible for the phenotypic heterogeneity of β-hemoglobinopathies.1,3-6 DNA methyltransferase 1 (DNMT1) is one such genetic modifier that regulates human γ-globin silencing epigenetically by maintaining promoter CpG methylation.7-9
The repressive effect of DNMT1 is functionally mediated by its N-terminal domain, which facilitates its interaction with numerous γ-globin regulators and is indispensable for the recruitment of DNMT1 to HBG promoters.9-11 Moreover, phosphorylation of serine residues within the N-terminal domain is an important factor in the catalytic activity and protein stability of DNMT1.12,13 Although clinical studies evaluating DMNT1 depletion have shown efficacy in upregulating HbF levels in patients with β-hemoglobinopathies,14-16 the association of DNMT1 mutations with β-thalassemia phenotypes and the underlying epigenetic mechanisms have yet to be demonstrated.
Here, we identified a novel missense mutation (c.2633G>A, S878F) in DNMT1 in patients with HbE/β-thalassemia, and uncovered a novel mechanism of this mutation in a DNMT1 germline mutant in direct epigenetic derepression of HBG, which in turn ameliorates the clinical severity of β-thalassemia.
Study design
A previously established cohort of 1142 Chinese β-thalassemia patients (supplemental Table 1, available on the Blood Web site) was recruited for molecular genetic analysis using genomic DNA.17 Target capture-based next-generation sequencing (NGS) was used to identify variants in a panel of candidate genetic modifiers associated with increased HbF, and the mutations were confirmed by Sanger sequencing. To uncover the mechanistic role of the novel S878F DNMT1 mutation in regulating the reactivation of γ-globin expression, the function of this mutation was analyzed through various in vitro assays using CD34+ cells from patients and HuDEP-2 cells, with the S878F mutation introduced by clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9). The F-cell pattern in patients’ blood and cultured erythroid cells was examined using immunofluorescence assays and flow cytometry analysis. Hematology assays, genetic tests, and DNA methylation assays were performed as previously described.5,17 Detailed protocols and information are described in supplemental Methods. Written informed consent was obtained from all participants and/or their family members.
Results and discussion
Using a target capture NGS approach for screening variants associated with altered HbF levels (supplemental Figures 1-2; supplemental Tables 2-5), we identified 10 potential HbF-regulating variants in IKZF2, CHD3, RAD21, and DNMT1 from a cohort of 1142 β-thalassemia patients. After further analysis, including prediction of well-known BCL11A variants influencing HbF levels, only a novel heterozygous missense mutation (c.2633G>A, p.Ser878Phe) in DNMT1 was found to be a positive modifier in 3 cases (supplemental Figure 2; supplemental Table 3). 17 A precedent for this mutation could not be found in the International HapMap Project (https://www.genome.gov/10001688/international-hapmap-project) or ChinaMAP (http://www.mbiobank.com/) databases (Figure 1A). The variant was further confirmed by Sanger sequencing, and the effect of the DNMT1 mutation in increasing HbF in individuals carrying the mutation from 2 Chinese pedigrees was determined. Among them, 3 patients (with β-thalassemia compound heterozygous mutations and this coinherited missense mutation) had high HbF (49.5%, 34.7%, and 32.5%, respectively) (Figure 1B; supplemental Table 6). Multiple alignment analysis suggested that this missense variant occurred at a highly conserved codon, changing a serine to phenylalanine at position 878 (S878F) in the first bromo-adjacent homology domain (BAH1) of DNMT118,19 (Figure 1C), with a predicted loss of 2 hydrogen bonds as essential structural elements (Figure 1D; supplemental Figure 3).
![A missense mutation in the DNMT1 gene ameliorates HbE/β-thalassemia. (A) Two respective cohorts, including 7968 individuals from southern China in the ChinaMap project (www.mbiobank.com) and a previously established cohort of 1142 southern Chinese β-thalassemia patients, were screened for the S878F mutation. The frequency and number of the S878F mutant allele in each cohort are shown. (B) Verification of the novel DNMT1 mutation and its correlation with HbF levels for 2 Chinese pedigrees. Representative Sanger sequencing chromatograms from 4 subjects are shown on the bottom, identifying a heterozygous DNMT1 missense mutation (c.2633G>A, p.Ser878Phe) in the probands of the 2 families. The genotypes of HBB gene mutations, DNMT1 mutation, and HbF levels (grams per deciliter) are indicated for each family member tested. Three positive cases with this mutation screened by a patient-based NGS approach are marked by red stars. (C) Conserved sequence analysis of the S878F mutation across vertebrate species. The structure of the protein encoded by DNMT1 is shown on the top. The S878F missense mutation (marked with a red star) occurred in the BAH1 domain, within the N-terminal regulatory domain. Below are enlarged graphs depicting a partial sequence alignment of the BAH1 domain around the position of this mutation, indicating a highly conserved amino acid sequence (shaded in light blue). (D) Left, prediction of hydrogen bond networks (broken black lines) formed by Ser878 (blue), Gln1293 (red), and Pro1320 (yellow) in WT DNMT1. Right, loss of hydrogen bonds in the S878F mutation DNMT1. The BAH1 domain is shaded in gold and the C-terminal catalytic domain is shaded in green. The image was generated using PyMOL (http://www.pymol.org/). (E-F) The effects of the S878F mutation on the levels of HbF (E) and age at onset of anemia (F) in individuals with HbE/β-thalassemia. Selected individuals had the same globin genotype category (HbE/β0 and αα/αα) and similar HbF-modulating genetic variants (KLF1WT/WT, rs368698783[GG], rs4671393 [GG or GA], or rs9399137 [TT or CT]). The data were obtained from a comparison of 3 cases with a DNMT1 mutation to 63 samples without this mutation in the control group, from a cohort of patients with 1142 β-thalassemia. Graphs depict the mutated group (blue column) and WT group (brown column), presented as the means ± standard error of the mean (SEM) (****P ≤ .0001; ***P ≤ .001). (G) Kaplan-Meier survival curves for the comparison of well-matched HbE/β0-thalassemia cases with or without the DNMT1 mutation. The sample-matched pair, 3 vs 63 patient cases with or without the S878F mutation are shown at the top of the chart. The 2 colored lines represent the 2 groups. Selection of samples is described in supplemental Tables 1 and 8. We used the log-rank test to compare the median age at first transfusion between the 2 groups (P = .027). DAF, derived allele frequency; PDB, Protein Data Bank.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/12/10.1182_blood.2020006425/1/m_bloodbld2020006425f1.png?Expires=1765885890&Signature=FxC8zNiTiRqHH85WEAdU8g3a3~tHktYEWSjWE~8KweaAivddpOoRZ~fZLhbtBN179V0-AKOc8X~kXzet0yCsvJfCbynooZXWAIQ0hmiWQg-U79GMSyhbRiLt2Pm9aQh-q0nz7P1fvBMhOpnj42VQgXxvAVV1Vfe2JE9TUZ1JJshDjLTmjQI8Ci8qFtaCNrIDwpod9r0qMicXOuKZj0Q4fXIgK3DV6ftGIDw4u1qZ4FPD6nzzqX3hAE-oOBdMmvE3NXFAPfL9IzQAld0oKJhQvrFCnS~w6wYB-clnjqA9V34tBgNIshQ2REVpcqPzCNwo5~4s56o1iKOo7Hypqbvwyw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A missense mutation in the DNMT1 gene ameliorates HbE/β-thalassemia. (A) Two respective cohorts, including 7968 individuals from southern China in the ChinaMap project (www.mbiobank.com) and a previously established cohort of 1142 southern Chinese β-thalassemia patients, were screened for the S878F mutation. The frequency and number of the S878F mutant allele in each cohort are shown. (B) Verification of the novel DNMT1 mutation and its correlation with HbF levels for 2 Chinese pedigrees. Representative Sanger sequencing chromatograms from 4 subjects are shown on the bottom, identifying a heterozygous DNMT1 missense mutation (c.2633G>A, p.Ser878Phe) in the probands of the 2 families. The genotypes of HBB gene mutations, DNMT1 mutation, and HbF levels (grams per deciliter) are indicated for each family member tested. Three positive cases with this mutation screened by a patient-based NGS approach are marked by red stars. (C) Conserved sequence analysis of the S878F mutation across vertebrate species. The structure of the protein encoded by DNMT1 is shown on the top. The S878F missense mutation (marked with a red star) occurred in the BAH1 domain, within the N-terminal regulatory domain. Below are enlarged graphs depicting a partial sequence alignment of the BAH1 domain around the position of this mutation, indicating a highly conserved amino acid sequence (shaded in light blue). (D) Left, prediction of hydrogen bond networks (broken black lines) formed by Ser878 (blue), Gln1293 (red), and Pro1320 (yellow) in WT DNMT1. Right, loss of hydrogen bonds in the S878F mutation DNMT1. The BAH1 domain is shaded in gold and the C-terminal catalytic domain is shaded in green. The image was generated using PyMOL (http://www.pymol.org/). (E-F) The effects of the S878F mutation on the levels of HbF (E) and age at onset of anemia (F) in individuals with HbE/β-thalassemia. Selected individuals had the same globin genotype category (HbE/β0 and αα/αα) and similar HbF-modulating genetic variants (KLF1WT/WT, rs368698783[GG], rs4671393 [GG or GA], or rs9399137 [TT or CT]). The data were obtained from a comparison of 3 cases with a DNMT1 mutation to 63 samples without this mutation in the control group, from a cohort of patients with 1142 β-thalassemia. Graphs depict the mutated group (blue column) and WT group (brown column), presented as the means ± standard error of the mean (SEM) (****P ≤ .0001; ***P ≤ .001). (G) Kaplan-Meier survival curves for the comparison of well-matched HbE/β0-thalassemia cases with or without the DNMT1 mutation. The sample-matched pair, 3 vs 63 patient cases with or without the S878F mutation are shown at the top of the chart. The 2 colored lines represent the 2 groups. Selection of samples is described in supplemental Tables 1 and 8. We used the log-rank test to compare the median age at first transfusion between the 2 groups (P = .027). DAF, derived allele frequency; PDB, Protein Data Bank.
A missense mutation in the DNMT1 gene ameliorates HbE/β-thalassemia. (A) Two respective cohorts, including 7968 individuals from southern China in the ChinaMap project (www.mbiobank.com) and a previously established cohort of 1142 southern Chinese β-thalassemia patients, were screened for the S878F mutation. The frequency and number of the S878F mutant allele in each cohort are shown. (B) Verification of the novel DNMT1 mutation and its correlation with HbF levels for 2 Chinese pedigrees. Representative Sanger sequencing chromatograms from 4 subjects are shown on the bottom, identifying a heterozygous DNMT1 missense mutation (c.2633G>A, p.Ser878Phe) in the probands of the 2 families. The genotypes of HBB gene mutations, DNMT1 mutation, and HbF levels (grams per deciliter) are indicated for each family member tested. Three positive cases with this mutation screened by a patient-based NGS approach are marked by red stars. (C) Conserved sequence analysis of the S878F mutation across vertebrate species. The structure of the protein encoded by DNMT1 is shown on the top. The S878F missense mutation (marked with a red star) occurred in the BAH1 domain, within the N-terminal regulatory domain. Below are enlarged graphs depicting a partial sequence alignment of the BAH1 domain around the position of this mutation, indicating a highly conserved amino acid sequence (shaded in light blue). (D) Left, prediction of hydrogen bond networks (broken black lines) formed by Ser878 (blue), Gln1293 (red), and Pro1320 (yellow) in WT DNMT1. Right, loss of hydrogen bonds in the S878F mutation DNMT1. The BAH1 domain is shaded in gold and the C-terminal catalytic domain is shaded in green. The image was generated using PyMOL (http://www.pymol.org/). (E-F) The effects of the S878F mutation on the levels of HbF (E) and age at onset of anemia (F) in individuals with HbE/β-thalassemia. Selected individuals had the same globin genotype category (HbE/β0 and αα/αα) and similar HbF-modulating genetic variants (KLF1WT/WT, rs368698783[GG], rs4671393 [GG or GA], or rs9399137 [TT or CT]). The data were obtained from a comparison of 3 cases with a DNMT1 mutation to 63 samples without this mutation in the control group, from a cohort of patients with 1142 β-thalassemia. Graphs depict the mutated group (blue column) and WT group (brown column), presented as the means ± standard error of the mean (SEM) (****P ≤ .0001; ***P ≤ .001). (G) Kaplan-Meier survival curves for the comparison of well-matched HbE/β0-thalassemia cases with or without the DNMT1 mutation. The sample-matched pair, 3 vs 63 patient cases with or without the S878F mutation are shown at the top of the chart. The 2 colored lines represent the 2 groups. Selection of samples is described in supplemental Tables 1 and 8. We used the log-rank test to compare the median age at first transfusion between the 2 groups (P = .027). DAF, derived allele frequency; PDB, Protein Data Bank.
Cox proportional hazards analysis demonstrated that S878F mutation is associated with a stronger effect on the clinical severity of β-thalassemia (hazard ratio = 0.230, P = .012; supplemental Table 7), measured by the age at first transfusion in 1142 β-thalassemia subjects. All 3 patients with S878F mutation were diagnosed with HbE/β0-thalassemia and exhibited significantly milder phenotypes than control group subjects without this mutation, as determined by case-control univariate analysis (Figure 1E-G; supplemental Table 8). Thus, the DNMT1 variant represents a novel genetic modifier, as evidenced by its association with phenotypes in β-thalassemia.
To investigate the precise mechanistic role of the S878F mutation in the reactivation of γ-globin, we performed functional analysis of this mutation in the HuDEP-2 cell line and primary CD34+ erythroid cells from 4 patients with HbE/β0-thalassemia (II-1 and II-3 in family 1, II-4 and II-5 in family 2). We first characterized the effect of the S878F mutation on interactions with coregulatory proteins, including a group of well-known erythroid transcription factors (BCL11A, SP1, GATA1, LRF, MYB, and TR2/TR4) and epigenetic coregulators (histone deacetylase 1/2 [HDAC1/2]) involved in macromolecular complexes that mediate HbF silencing.9-12,19,20 S878F mutation dramatically decreased the interaction of DNMT1 with BCL11A, GATA1, and HDAC1/2, as examined by coimmunoprecipitation using either target protein-specific antibodies (Figure 2A) or anti-DNMT1 antibody (Figure 2B) in CD34+ erythroid cells with mutated compared with wild-type (WT) DNMT1. This analysis suggests that the mutated BAH1 domain obstructs the formation of macromolecular complexes via protein-protein interactions.19,20 These results are consistent with those observed in HuDEP-2 cells (supplemental Figure 4A-B). Collectively, our results suggest that the structure of the mutant S878F protein disturbs the protein-protein interactions between DNMT1 and the coregulators.

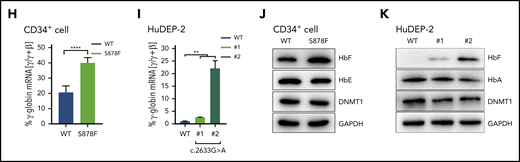
The S878F mutation leads to hypomethylation-mediated reactivation of γ-globin. (A-B) Analysis of protein-protein interaction by coimmunoprecipitation in CD34+ cells with or without S878F mutation. Proteins copurified from CD34+ cell nuclear extracts were analyzed by western blotting (WB) with specific antibodies against target proteins (A) or anti-DNMT1 antibody (B) using isotype-matched immunoglobulin G (IgG) antibody as a negative control. All of the inputs are 5%. The WT and S878F samples are marked as WT (left column) or S878F (right column) at the top of the graphs. Each of the different target proteins examined are indicated at the right of the strips. LRF protein could be not detected in either WT or S878F cells using anti-DNMT1 antibodies, implying that an indirect interaction may exist between DNMT1 and LRF. (C) Analysis of the phosphorylation status of the serine residue at position 878 of DNMT1 in nuclear lysates of HuDEP-2 (left) or CD34+ cells (right). HuDEP-2 cells were transfected with WT or S878F DNMT1 constructs. Proteins copurified from constructed HuDEP-2 cells or CD34+ cells were analyzed by WB with the antiphosphorylated form, BAH1 Ser878 (pDNMTS878) antibody, using anti-DNMT1 and anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies as controls. (D) Analysis of the protein degradation of DNMT1 in HuDEP-2 cells transfected with vectors encoding enhanced green fluorescent protein (EGFP)-tagged full-length WT (left) or mutated DNMT1 (right). Seventy-two hours after transfection, cells were treated with cycloheximide and collected at 4 time points (0 hours, 4 hours, 6 hours, and 8 hours). DNMT1 was detected by WB using anti-green fluorescent protein (GFP) antibody and GAPDH as a loading control. The degradation curve graph is shown at the bottom. (E) Assay of DNA methyltransferase activity in nuclear extracts of HuDEP-2 cells transfected with vectors encoding EGFP-tagged full-length WT or mutation DNMT1. DNA methyltransferase activity was determined using a hemi-methylated DNA-trapping assay. (F) ChIP-qPCR analysis of the relative enrichment of DNMT1 at the γ-globin promoter in CD34+ cells. The GAPDH promoter served as positive control (+ctrl) and the β-globin promoter served as negative control (−ctrl). Results were normalized to the positive control. (G) Effects of the S878F mutation on the HBG promoter methylation level were examined by bisulfite-sequencing method in CD34+ cells. Graphic location of analyzed CpG sites in the promoter regions with sequence homology in 2 HBG genes (Gγ and Aγ) is shown in the left upper region. Sequence variations of 2 subjects with or without the S878F mutation are shown at the left lower section of the sequencing chromatograph. Right panel, methylation levels at 6 CpG sites (−162, −53, −50, +6, +17, and +50) obtained from tested cases with or without the S878F mutation. (H-K) Quantitative measurement of HBG mRNA expression by qPCR (H-I) and HbF production by WB (J-K) in CD34+ cells and CRISPR/Cas9-edited S878F mutant HuDEP-2 cells. All CD34+ cells were collected from HbE/β0-thalassemia patients. Data in the column graphs (E-I) from at least 2 independent experiments are presented as the means ± SEM (****P ≤ .0001; ***P ≤ .001; **P ≤ .01). IP, immunoprecipitation; LRF, leukemia/lymphoma-related factor; mCpG, methylated CpG.
The S878F mutation leads to hypomethylation-mediated reactivation of γ-globin. (A-B) Analysis of protein-protein interaction by coimmunoprecipitation in CD34+ cells with or without S878F mutation. Proteins copurified from CD34+ cell nuclear extracts were analyzed by western blotting (WB) with specific antibodies against target proteins (A) or anti-DNMT1 antibody (B) using isotype-matched immunoglobulin G (IgG) antibody as a negative control. All of the inputs are 5%. The WT and S878F samples are marked as WT (left column) or S878F (right column) at the top of the graphs. Each of the different target proteins examined are indicated at the right of the strips. LRF protein could be not detected in either WT or S878F cells using anti-DNMT1 antibodies, implying that an indirect interaction may exist between DNMT1 and LRF. (C) Analysis of the phosphorylation status of the serine residue at position 878 of DNMT1 in nuclear lysates of HuDEP-2 (left) or CD34+ cells (right). HuDEP-2 cells were transfected with WT or S878F DNMT1 constructs. Proteins copurified from constructed HuDEP-2 cells or CD34+ cells were analyzed by WB with the antiphosphorylated form, BAH1 Ser878 (pDNMTS878) antibody, using anti-DNMT1 and anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies as controls. (D) Analysis of the protein degradation of DNMT1 in HuDEP-2 cells transfected with vectors encoding enhanced green fluorescent protein (EGFP)-tagged full-length WT (left) or mutated DNMT1 (right). Seventy-two hours after transfection, cells were treated with cycloheximide and collected at 4 time points (0 hours, 4 hours, 6 hours, and 8 hours). DNMT1 was detected by WB using anti-green fluorescent protein (GFP) antibody and GAPDH as a loading control. The degradation curve graph is shown at the bottom. (E) Assay of DNA methyltransferase activity in nuclear extracts of HuDEP-2 cells transfected with vectors encoding EGFP-tagged full-length WT or mutation DNMT1. DNA methyltransferase activity was determined using a hemi-methylated DNA-trapping assay. (F) ChIP-qPCR analysis of the relative enrichment of DNMT1 at the γ-globin promoter in CD34+ cells. The GAPDH promoter served as positive control (+ctrl) and the β-globin promoter served as negative control (−ctrl). Results were normalized to the positive control. (G) Effects of the S878F mutation on the HBG promoter methylation level were examined by bisulfite-sequencing method in CD34+ cells. Graphic location of analyzed CpG sites in the promoter regions with sequence homology in 2 HBG genes (Gγ and Aγ) is shown in the left upper region. Sequence variations of 2 subjects with or without the S878F mutation are shown at the left lower section of the sequencing chromatograph. Right panel, methylation levels at 6 CpG sites (−162, −53, −50, +6, +17, and +50) obtained from tested cases with or without the S878F mutation. (H-K) Quantitative measurement of HBG mRNA expression by qPCR (H-I) and HbF production by WB (J-K) in CD34+ cells and CRISPR/Cas9-edited S878F mutant HuDEP-2 cells. All CD34+ cells were collected from HbE/β0-thalassemia patients. Data in the column graphs (E-I) from at least 2 independent experiments are presented as the means ± SEM (****P ≤ .0001; ***P ≤ .001; **P ≤ .01). IP, immunoprecipitation; LRF, leukemia/lymphoma-related factor; mCpG, methylated CpG.
Using a specific antibody against the phosphorylated form of Ser878 in DNMT1, we demonstrated abrogation of DNMT1 phosphorylation at position 878 of the mutant S878F protein in HuDEP-2 cells (Figure 2C, left panel) and CD34+ erythroid cells (Figure 2C, right panel). We also observed that the mutant S878F protein displayed a faster degradation rate than its WT counterpart (Figure 2D), and was correlated with significantly decreased methyltransferase activity of DNMT1 in HuDEP-2 cells (Figure 2E). These results suggest that DNMT1 mutation leads to impairment of both its structural stability and catalytic activity, which is similar to results from previous studies of phosphorylation at Ser154 of DNMT1.13 However, these observations were not replicated in the HEK-293T human renal epithelial cell line when transfected with either WT DNMT1 or mutated DNMT1 (S878F) (supplemental Figure 5), suggesting that phosphorylation of DNMT1 at residue Ser878 or BAH1 domain–mediated interactions may be regulated in a tissue- or developmental-stage restricted manner.
We further evaluated the impact of S878F mutation on HBG promoter methylation and γ-globin expression both in CD34+ cells from patients and CRISPR/Cas9-edited S878F mutant HuDEP-2 cells (supplemental Figure 6; supplemental Table 10). Chromatin immunoprecipitation (ChIP) quantitative polymerase chain reaction (qPCR) showed that S878F mutation significantly reduced the accumulation of DNMT1 at the HBG promoter,7,21 but no changes were observed in the enrichment of GATA1 and HDAC2 between the WT and mutated CD34+ cells (Figure 2F; supplemental Figure 7), indicating the involvement of DNMT1 in this regulation. We next investigated the DNA methylation status at the HBG promoter, and the S878F mutant exhibited a high degree of hypomethylation at 6 CpG sites (−162, −53, −50, +6, +17, and +50) in HBG promoter regions (Figure 2G; supplemental Figure 8), but no changes were observed in global methylation levels between the WT and mutated proteins in either CD34+ or HuDEP-2 cells (supplemental Figure 9), suggesting a narrower set of demethylation changes confined to the γ-globin genes and their active participation in the reactivation of γ-globin. We also determined that γ-globin expression was increased at both the messenger RNA (mRNA) and protein levels in CD34+ and HuDEP-2 cells (Figure 2H-K; supplemental Figure 10), and the effect of S878F mutation was heterocellular, as a result of an increased number of HbF+ cells (F cells) (supplemental Figure 11). Furthermore, significant changes were not observed in erythroid differentiation kinetics in CD34+ cells from patients with HbE/β0-thalassemia (supplemental Figure 12).
Our results not only suggest that γ-globin gene methylation is reduced either by decreased DNMT1 binding to HBG promoters (Figure 2F; supplemental Figure 7) or impaired catalytic activity (Figure 2D-E), but also that S878F mutation attenuated DNMT1 interactions with BCL11A, GATA1, and HDAC1/2 (Figure 2A-B). Taken together, we propose that the reactivation of γ-globin induced by DNMT1 S87F mutation constitutes a mechanism for reduced γ-globin gene methylation in combination with reduced recruitment of a corepressor complex.
In summary, we identified a DNMT1 variant (S878F) as a novel genetic modifier responsible for high HbF production by epigenetically derepressing γ-globin expression and thereby ameliorating β-thalassemia severity. Our findings suggest that the BAH1 domain of DNMT1 harboring a serine at position 878 maintains DNA methylation. This mutation occurs naturally in a benign genetic condition, which could potentially be recapitulated in primary erythroid cells by Cas9 or base-editor technology22 to treat β-hemoglobinopathies.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all those who participated in this study. They are particularly grateful to Dali Li from East China Normal University, and Xingxu Huang Shanghai Tech University for their timely help with editing erythroid cells. The authors also thank Wei Liao from the 923rd Hospital of the People’s Liberation Army for assistance in collecting patient samples; Fu Xiong, Qiyang Li, Jiaqiong Lin, Jie Bai, Zhongju Wang, and Xuan Shang from Southern Medical University and Peng Xu from Soochow University for valuable advice and comments on this work; and Mingli Xu and Zhunqiang Zhong from Southern Medical University for assistance in conducting technical work.
This work was supported by grants from the National Key Research and Development Program of China (2018YFA0507800 and 2018YFA0507803) and the National Natural Science Foundation of China (31671314 and 31871265).
Authorship
Contribution: Y.G., Y.Y., and X.X. designed the study; Y.G., Y.Z., J.H., Liren Wang, W.Y., and C.S. performed the experiments and analyzed the data; Y.L., T.Y., and Li Wang collected samples and clinical data; X.Z. performed clinical classifications and molecular diagnosis and managed β-thalassemia patients; Q.Z., Y.Y., and J.Z. analyzed the data; Y.G., Q.Z., and X.X. wrote the manuscript; X.X. supervised the study; and all the authors read and approved the contents of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xiangmin Xu, Department of Medical Genetics, School of Basic Medical Science, Southern Medical University, Guangzhou, 510515, Guangdong, China; e-mail: gzxuzm@pub.guangzhou.gd.cn or xixm@smu.edu.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal