

TO THE EDITOR:
In sickle cell disease (SCD), a single base mutation in HBB generates a hemoglobin variant (sickle hemoglobin [HbS]), which polymerizes within the cell upon deoxygenation and produces the characteristic sickle-shaped erythrocyte that defines the disease.1 Although it has long been understood that there is a delay between deoxygenation and the initiation of sickling that is strikingly dependent on the intracellular concentration of HbS,2 directly targeting Hb concentration is an underinvestigated therapeutic approach. Case reports and small studies have reported associations between iron deficiency, decreased mean corpuscular Hb concentration (MCHC), and improvement in several clinical parameters, including decreased sickling and hemolysis, increased red blood cell (RBC) lifespan, decreased hospitalization for pain crises, and improved leg ulcer healing.3-5 Additional support for iron restriction as a therapeutic approach is found in a murine transplant model of SCD, in which mice with iron deficiency (consequent to an inactivating mutation of the intestinal Hif-2α gene) had better RBC survival and less severe anemia when compared with iron-sufficient wild-type mice likewise transplanted with SCD marrow.6
Collectively, these data led us to hypothesize that manipulation of Hb concentration by an alternate approach such as dietary iron restriction might produce a similar decrease in Hb concentration with beneficial effects on sickling and hemolysis. To test this hypothesis, the effects of modest dietary iron restriction on hematologic status were investigated in the Townes murine model7 of SCD. Sickle trait mice (HbAS) or SCD (HbSS) mice were weaned onto either an iron-restricted diet containing 20 parts per million (ppm) iron or an otherwise identical iron-adequate diet containing 48 ppm iron. HbSS mice with iron restriction demonstrated improved hematologic parameters. Mice that had an iron-restricted diet had decreased serum iron concentrations (Figure 1A), decreased MCHC (Figure 1B), increased number of circulating RBCs (Figure 1C), and improved hematocrit (Figure 1D) compared with mice fed the standard diet. The decrease in MCHC reflected the decreased, albeit not statistically significant, mean cellular Hb (supplemental Figure 1A, available on the Blood Web site) and increased mean corpuscular volume (supplemental Figure 1B). No statistical differences were observed in Hb concentration (Figure 1E) or reticulocyte count (Figure 1F) as a function of diet in the HbSS mice. The improved hematocrit translated to improved tissue oxygenation as evidenced by decreased kidney erythropoietin expression in the iron-restricted HbSS mice (supplemental Figure 2A). Trait mice did not differ in any of these parameters as a function of dietary iron.
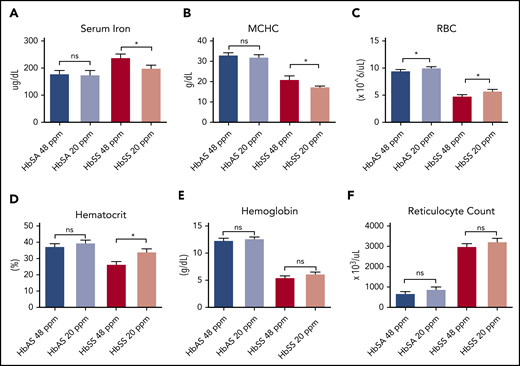
Iron restriction improves hematological parameters in Townes sickle mice. (A-E) Townes HbSS and trait HbAS mice weaned and maintained on either a 48- or 20-ppm iron diet for 8-10 weeks. Then serum iron (A), mean corpuscular hemoglobin concentrations (B), red blood cell counts (C), hematocrit (D), hemoglobin concentration (E), and reticulocyte count (F) were measured; n = 6-13 mice per group. Data represented as means ± standard error of the mean. *P < .05; ***P < .005; ****P < .001. ns, nonsignificant.
Iron restriction improves hematological parameters in Townes sickle mice. (A-E) Townes HbSS and trait HbAS mice weaned and maintained on either a 48- or 20-ppm iron diet for 8-10 weeks. Then serum iron (A), mean corpuscular hemoglobin concentrations (B), red blood cell counts (C), hematocrit (D), hemoglobin concentration (E), and reticulocyte count (F) were measured; n = 6-13 mice per group. Data represented as means ± standard error of the mean. *P < .05; ***P < .005; ****P < .001. ns, nonsignificant.
We next investigated whether the lower erythrocyte Hb concentration in the iron-restricted HbSS mice affects hypoxia-induced sickling. We used oxygen gradient ektacytometry (oxygenscan) to measure the deformability of RBCs as a function of progressive ex vivo decreases in oxygen tension.8 We observed that RBCs from the iron-restricted HbSS mice have a lower point of sickling, that is, the oxygen tension at which a 5% decrease in deformability becomes apparent (Figure 2A). The severity of sickling is reflected in the “ΔEI” (ie, the difference between the maximum elongation index [EI max], measured when the cells are fully oxygenated, and the minimum elongation index [EI min], measured when the cells are maximally deoxygenated). Dietary iron restriction improved the ΔEI for the HbSS mice (Figure 2B). This improvement was not the result of changes in deformability, because there was no significant effect of iron diet on the elongation of the fully oxygenated erythrocytes (EI max; Figure 2D). However, cells from HbSS mice receiving the iron-restricted diet had significantly higher EI min (Figure 2C) compared with those on the standard iron diet. These findings demonstrate a beneficial effect of iron restriction on hypoxia-induced sickling.
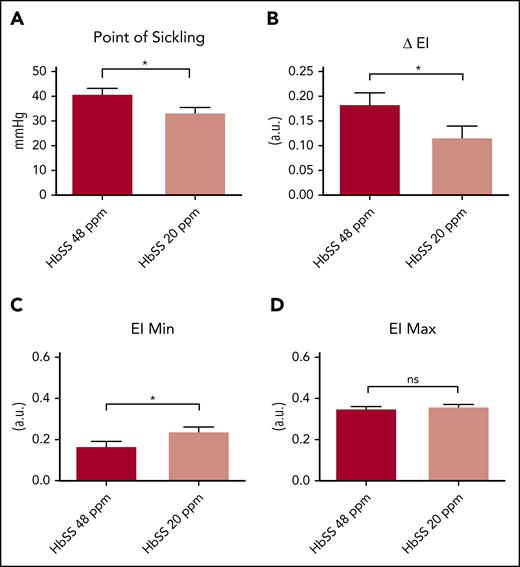
Iron restriction attenuates hypoxia induced sickling. (A) Point of sickling of RBCs from Townes HbSS mice fed a 20- or 48-ppm iron diet. (B-D) Difference in EI (B), EI min (C), and EI max (D) in HbSS mice fed a 20- or 48-ppm iron diet. For panel A, n = 9-13 mice per group. For panels B-D, n = 10-13 mice per group. Data represented as means ± standard error of the mean. *P < .05; ****P < .001. ns, nonsignificant.
Iron restriction attenuates hypoxia induced sickling. (A) Point of sickling of RBCs from Townes HbSS mice fed a 20- or 48-ppm iron diet. (B-D) Difference in EI (B), EI min (C), and EI max (D) in HbSS mice fed a 20- or 48-ppm iron diet. For panel A, n = 9-13 mice per group. For panels B-D, n = 10-13 mice per group. Data represented as means ± standard error of the mean. *P < .05; ****P < .001. ns, nonsignificant.
In accordance with the decreased propensity to sickle, the endothelial cell activation marker serum soluble vascular cell adhesion molecule-1 (sVCAM-1)9 is decreased in the iron-restricted HbSS mice (supplemental Figure 2B). Serum unconjugated (indirect) bilirubin is likewise lower in the HbSS mice fed the 20-ppm diet compared with those fed the standard diet (supplemental Figure 2C), suggesting that dietary iron restriction decreases hemolysis.10 Kidney iron concentration, an identified marker of chronic hemolysis, is also lower in the iron-restricted HbSS mice, which provides additional support for decreased RBC rupture in iron-restricted mice (supplemental Figure 3A). Dietary iron restriction decreases splenic iron concentration in both HbAS and HbSS mice (supplemental Figure 3B) compared with a standard iron diet. It is worth noting that although the splenic iron is higher per gram of tissue in the HbAS mice fed a standard iron diet, profound splenomegaly is characteristic of the HbSS mice, and thus total spleen iron is markedly increased in the HbSS mice. Liver iron load, higher in HbSS mice than in HbAS mice, decreases substantially in the HbSS mice with iron restriction (supplemental Figure 3C). Likely in response to an enhanced erythroid signal,11 the iron regulatory hormone hepcidin is lower in the HbSS mice that received a standard iron diet than the HbSA mice. Dietary iron restriction lowers hepcidin expression further in the HbSS mice (supplemental Figure 3D). Although the modest change in dietary iron has limited effects in the HbAS mice, it is adequate for substantially mitigating tissue iron loading and decreasing hepcidin expression in the HbSS mice. These findings support decreased chronic hemolysis, improved tissue oxygenation, and improved tissue iron status in the iron-restricted HbSS mice.
On the basis of these observations, we speculate that modest iron restriction may decrease the clinical complications arising from both sickling and hemolysis in SCD. Dietary iron restriction is unlikely to be tractable in patients. Future directions will explore therapeutic approaches beyond diet to restrict iron availability to the erythron in SCD. Several potential therapeutics that modulate iron via the hepcidin-ferroportin axis, including minihepcidins,12 an oral ferroportin inhibitor,13 and antisense oligonucleotides targeting the gene that encodes the serine protease Matriptase-214 are currently under development. Murine models of SCD provide a model system for preclinical characterization of these and other therapeutic approaches that will serve to inform future clinical trials of iron restriction in SCD patients.
For original data, please contact Nermi L. Parrow via e-mail at nermi.parrow@health.slu.ed.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Monica Rao and Awais Paracha for technical assistance.
This work was supported, in part, by grants from the National Institutes of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK-R01DK095112) (R.E.F.), the NIH/Clinical and Translational Science Awards Program (CTSA-UL1 TR002345) (N.L.P. and R.E.F.), by the Saint Louis University School of Medicine President’s Research Fund (R.E.F.), and by the Intramural Research Programs of the NIH/National Institute of Diabetes and Digestive and Kidney Diseases and the NIH/National Heart, Lung, and Blood Institute.
The opinions expressed herein are the sole responsibility of the authors and do not necessarily represent the official views of the NIH.
Authorship
Contribution: R.E.F., J.F.T., C.F., S.L.T., and N.L.P. conceived the study; R.E.F., N.L.P., P.-C.V., and M.L. chose the methodology; N.L.P., P.-C.V., N.A.G., F.A., S.B., R.W., M.L., and R.E.F. conducted the investigation; R.E.F., N.L.P., and P.-C.V. performed the formal analysis; R.E.F., N.L.P., and P.-C.V. wrote the original draft of the manuscript; R.E.F., M.L., C.F., and J.F.T. acquired the funding; and R.E.F. supervised the project.
Conflict-of-interest disclosure: R.E.F. is a member of the Scientific Advisory Boards of Protagonist Therapeutics and Silence Therapeutics and receives research funding from Ultragenyx. The remaining authors declare no competing financial interests.
Correspondence: Nermi L. Parrow, Saint Louis University School of Medicine, 1100 S Grand Blvd, St. Louis, MO 63104; e-mail: nermi.parrow@health.slu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal