

Key Points
SRP54 mutations induce CN and its syndromic form, SDS.
Impaired unconventional splicing of XBP1 is a key player in SRP54-mediated disease.

Abstract
Heterozygous de novo missense variants of SRP54 were recently identified in patients with congenital neutropenia (CN) who display symptoms that overlap with Shwachman-Diamond syndrome (SDS). Here, we investigate srp54 knockout zebrafish as the first in vivo model of SRP54 deficiency. srp54−/− zebrafish experience embryonic lethality and display multisystemic developmental defects along with severe neutropenia. In contrast, srp54+/− zebrafish are viable, fertile, and show only mild neutropenia. Interestingly, injection of human SRP54 messenger RNAs (mRNAs) that carry mutations observed in patients (T115A, T117Δ, and G226E) aggravated neutropenia and induced pancreatic defects in srp54+/− fish, mimicking the corresponding human clinical phenotypes. These data suggest that the various phenotypes observed in patients may be a result of mutation-specific dominant-negative effects on the functionality of the residual wild-type SRP54 protein. Overexpression of mutated SRP54 also consistently induced neutropenia in wild-type fish and impaired the granulocytic maturation of human promyelocytic HL-60 cells and healthy cord blood–derived CD34+ hematopoietic stem and progenitor cells. Mechanistically, srp54-mutant fish and human cells show impaired unconventional splicing of the transcription factor X-box binding protein 1 (Xbp1). Moreover, xbp1 morphants recapitulate phenotypes observed in srp54 deficiency and, importantly, injection of spliced, but not unspliced, xbp1 mRNA rescues neutropenia in srp54+/− zebrafish. Together, these data indicate that SRP54 is critical for the development of various tissues, with neutrophils reacting most sensitively to the loss of SRP54. The heterogenic phenotypes observed in patients that range from mild CN to SDS-like disease may be the result of different dominant-negative effects of mutated SRP54 proteins on downstream XBP1 splicing, which represents a potential therapeutic target.
Introduction
Congenital neutropenias (CNs) encompass a group of heterogeneous inherited disorders characterized by a reduction of neutrophil counts, recurrent infections and an elevated risk for the development of myeloid malignancies. Depending on the underlying genetic cause, additional defects may occur, such as exocrine pancreatic insufficiency in patients with Shwachman-Diamond syndrome (SDS)1,2-5 or cardiac and urogenital malformations in patients harboring G6PC3 mutations.4,6-8
Mutations in the ELANE and SBDS genes are among the most common genetic aberrations in CN and its syndromic form, SDS, respectively.4,9 In past years, research increasingly focused on identifying other hitherto unknown mutations in CN patients without established genetic background, eventually leading to the discovery of more than 20 genetic lesions associated with CN.4,10,11 By performing classical exome sequencing on SBDS-negative SDS-like patients, we recently identified mutations in the gene encoding signal recognition particle 54 (SRP54) as a novel cause of the disease.1 SRP54 is a part of the signal recognition particle (SRP) ribonucleoprotein complex that mediates the cotranslational targeting of nascent proteins to the endoplasmic reticulum (ER).1,12 Interestingly, heterozygous SRP54 mutations are associated with a wide range of phenotypic diseases, from mild CN to severe SDS. In our initial study, 2 of the 3 identified novel mutations were found in patients with SDS-like features (p.T115A, p.G226E), while the third was found in a patient suffering from isolated CN (p.T117Δ). In accordance with our results, another study noted that the p.T117Δ mutation commonly occurred in CN.13,14
Here, we show that the heterogenic clinical phenotypes observed with different SRP54 lesions can be explained by various mutation-specific dominant effects of the mutated protein on the residual wild-type (WT) SRP54 protein. Furthermore, we provide in-depth characterization of an srp54 zebrafish knockout (KO) mutant and demonstrate that SRP54 mutations impair granulocytic maturation by hampering the unconventional splicing of the transcription factor XBP1, which ultimately leads to unresolved ER stress and blockade of terminal neutrophil differentiation.
Methods
Zebrafish husbandry and genetic strains
Zebrafish were bred and maintained at 28°C as described.15 Staging was performed during hours postfertilization (hpf) and according to Federation of European Laboratory Animal Science Associations and Swiss federal law guidelines.16 The following lines were used in this study: WT Tübingen strains, Tg(mpo:eGFP), and srp54sa11820.17,18 Kaplan-Meier survival analysis was performed on srp54+/−, srp54−/−, and WT siblings. Genotyping was performed on whole embryos, tail clips, or dissociated cells according to standard protocols.19 Polymerase chain reaction (PCR) was performed using primers spanning the sa11820 mutation (forward: 5′-TTTGCAGATGCAGATGCACTTCTTAAAAT-3′; reverse: 5′-CCATGCAATGACGTTTTGTT-3′). Sanger sequencing was performed using the already mentioned reverse primer, and data were analyzed by using Lasergene SeqMan Pro software (DNASTAR, Madison, WI).
Messenger RNA injections
Capped messenger RNA (mRNA) of human SRP54 mutants was produced as described previously.1 To generate capped mRNA of zebrafish spliced xbp1 (xbp1s) and unspliced xbp1 (xbp1u), total RNA was extracted from ∼50 pooled WT embryos according to Peterson and Freeman.20 Reverse transcription (RT) was performed using Multiscribe Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA). PCR using Phusion Polymerase (Thermo Fisher Scientific) was performed on complementary DNA (cDNA) (xbp1u-forward: 5′-CCATCGATTCGAATTATGGTCGTAGTTACAGCAGGGAC-3′ and xbp1u-reverse: 5′-GAGAGGCCTTGAATTTCAGTTCATTAAGGGCTTCCAGCT-3′; xbp1s-forward: 5′-CCATCGATTCGAATTATGGTCGTAGTTACAGCAGGGAC-3′ and xbp1s-reverse: 5′-GAGAGGCCTTGAATTTCAGACGCTAATCAGTTGGGG G-3′) and cloned into the PCS2+ vector by InFusion cloning (Takara Bio Europe, Saint-Germain-en-Laye, France). Capped mRNA was generated with the AmpliCap SP6 High Yield Message Maker Kit (CELLSCRIPT, Madison, WI) and purified with ammonium acetate according to standard protocols.
Embryos were injected at the single-cell stage using mRNA concentrations of 50 ng/µL. Phenol red (0.05%) (Sigma-Aldrich, St. Louis, MO) was added as an injection tracer. Embryos were raised to appropriate stages and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for further analyses. For rescue experiments, human WT and mutated SRP54 mRNAs were generated as described.1
Injection of morpholino oligonucleotides
An xbp1 splice morpholino oligonucleotide (MO) that skipped exon 2 to prevent pre-mRNA splicing was synthetized by Gene Tools (Gene Tools, Philomath, OR): ACAATGGTCAAAGTACCTCCAGCTC. The MO was validated by reverse transcription polymerase chain reaction (RT-PCR). The primers were designed in the exons before and after exon 2, respectively (forward: CCTCTGGACCACCACTGAGA; reverse: CCAGTCTCTGTCTCAGCTCC). Embryos were injected at the single-cell stage. Phenol red (0.05%) (Sigma-Aldrich) was added as an injection tracer. Embryos were raised to appropriate stages and fixed in 4% paraformaldehyde in PBS for further analyses.
Whole-mount in situ hybridization and flow cytometry
Whole-mount in situ hybridization (WISH) was performed as described previously.1,21 Positive cells from WISH were semi-automatically counted using Fiji software,22 which was also used to measure changes in pancreas size. For histopathologic analyses, fish were fixed according to standard procedures. Fixed embryos were then automatically prepared and subsequently paraffin embedded using the Tissue Processor TPC15 and the TBS88 Paraffin Embedding System (Medite GmbH, Burgdorf, Germany) and cut into 5-μm thick slices with a Microtom HM430 (Thermo Fisher Scientific,). Photographs were taken with a Leica DM 2000 LED microscope.
For flow cytometry, single dechorionated transgenic embryos were dissociated into single cells as previously described1 in a 96-well plate. The number of fluorescence-labeled cells was then determined on a Beckman Coulter CytoFlex flow cytometer, and data were analyzed using FlowJo software (FlowJo, Ashland, OR). Remaining cells were used for genotyping as described above.
Neutrophil migration assay
Tg(srp54+/−; mpo:GFP) zebrafish were crossed to Tg(srp54+/−) zebrafish, and their progeny were raised to 48 hpf. At 48 hpf, tail fin wounding was performed as described previously.1 mpo+ cells were counted automatically using Fiji software.22 Imaging was performed with a Leica SP5-II-MATRIX microscope.
Isolation of whole kidney marrow (WKM) and flow cytometric analysis
Neutrophil differentiation assay
cDNA encoding for human SRP54 (WT) or its derived mutant forms p.T115A, p.T117Δ, and p.G226E were PCR amplified from pCS2 source vectors1 (primer forward: 5′-GCGAGATCGATCACCATGGTTCTAGCAGACCTTGG-3′; reverse: 5′-CTGACATCGATTTACATATTATTGAATCCCA-3′) and subcloned into a spleen focus-forming virus overexpression vector using the ClaI site. Sequence-verified plasmids were lentivirally integrated into promyeloid HL-60 cells (CCL-240; American Type Culture Collection) according to standard procedures and selected for cotransduced internal ribosome entry site/green fluorescent protein (IRES/GFP) by fluorescence-activated cell sorting. Resultant SRP54 overexpression cells were propagated in RPMI medium supplemented with 10% fetal calf serum and antibiotics, and granulocytic cell differentiation was induced with 1 µM all-trans retinoic acid (ATRA) (Sigma-Aldrich), as described.25,26 On day 6, nuclear lobulation was quantified as an indicator of neutrophilic differentiation27 in hematoxylin and eosin–stained cytospins (microscope: Leica DM 2000 LED, 40× objective; LAS EZ software), and granulocytic differentiation was further assessed by respective surface staining (allophycocyanin [APC]/Cy7 anti-human CD11b, BioLegend, San Diego, CA; APC mouse anti-human CD15, BD Biosciences, San Jose, CA; and phycoerythrin [PE]-Cy5 mouse anti-human CD16, BD Biosciences) and flow cytometry (Fortessa; BD, Franklin Lakes, NJ)
Isolation and differentiation of CD34+ hematopoietic stem and progenitor cells
Human cord blood samples from healthy newborn babies of both sexes were collected at the University Hospital Basel upon availability. Mononuclear cells were enriched by using a Ficoll gradient (Biocoll; Merck Millipore, Darmstadt, Germany), and CD34+ progenitors were purified by using a magnetically activated cell sorter. Purified cells were cultured in Iscove modified Dulbecco medium supplemented with interleukin-3 and stem cell factor as described by Gupta et al.28 After 1 day, the cells were lentivirally transduced with the previously described plasmids overexpressing either WT or p.G226E SRP54 according to standard protocols. Subsequently, differentiation of transduced cells was carried out as described by Gupta et al.28 Maturation was assessed by flow cytometric analyses of CD11b surface expression on cells that are double-positive for CD15 and CD16 (PE anti-human CD11b, BioLegend; APC mouse anti-human CD15, BD Biosciences; PE-Cy5 mouse anti-human CD16, BD Biosciences).
Tunicamycin treatment
Tg(srp54+/−; mpo:GFP) zebrafish were crossed with Tg(srp54+/−) fish, and their progeny were raised to 24 hpf. At 24 hpf, embryos were treated with 2 µg/mL Tunicamycin (Tm; Sigma-Aldrich) and incubated for 24 hours.29 At 48 hpf, zebrafish embryos were dissociated into single cells as previously described.1 To genotype the fish, 10% of the whole-cell suspension was used, and the remaining 90% was used either as input for quantitative RT-PCR (qRT-PCR) or for flow cytometric analysis of neutrophil counts. Transduced cells from the HL-60 cell line were treated with 5 μg/mL Tm for 5 hours, washed once with PBS, and then analyzed by qRT-PCR.
qRT-PCR
Total RNA was extracted from cell suspensions of dissociated single embryos and transduced HL-60 cells using the PicoPure RNA Isolation Kit (Thermo Fisher Scientific). cDNA was generated by RT using Multiscribe Reverse Transcriptase (Thermo Fisher Scientific) and later diluted 1:10 and used as input for real-time PCR. Real-time PCR was performed on an Applied Biosystems 7500 Real-Time PCR System (Thermo Fisher Scientific) using FastStart Universal SYBR Green Master (Rox) (Sigma-Aldrich) (xbp1s-forward: 5′-TGTTGCGAGACAAGA-3′ and xbp1s-reverse: 5′-CCTGCACCTGCTGCGGACT-3′ [for atf4, bip, and chop, see Vacaru et al30 ; for XBP1s and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), see Yoon et al31 ]).
Immunoblotting
Transduced HL-60 cells were disrupted in 1× Lysis Buffer (Cell Signaling, Danvers, MA) supplemented with Protease/Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) and cleared by centrifugation (15 minutes at 21 000 relative centrifugal force at 4°C). Cleared protein lysates were denatured with 4× Laemmli buffer.
In all, 5-10 zebrafish embryos (48 hpf) were dechorionated, deyolked, and homogenized with a microfuge pestle in sodium dodecyl sulfate sample buffer according to Westerfield.32 HL-60 cells or zebrafish cell lysates were separated over 12% bis-acrylamide gels (BioRad, Hercules, CA) by discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membrane (Amersham and GE Healthcare Life Sciences, Chalfont St. Giles, United Kingdom) in a semi-dry blotting apparatus (Trans-Blot Turbo, BioRad). Membranes were blocked with 10% w/v nonfat dry milk (Cell Signaling Technology, Danvers, MA) diluted in Tris-buffered saline 0.1% Tween-20 (Sigma-Aldrich), and proteins were stained with the following primary antibodies: anti-SRP54 (GTX115041; GeneTex, Irvine, CA) and anti-GAPDH (#5174, Cell Signaling Technologies, used for human samples; #60004-1-Ig; Proteintech Group, Rosemont, IL, used for zebrafish samples). Proteins were detected by an electrochemiluminescence reaction involving the horseradish peroxidase–linked anti-rabbit (#7074) or horseradish peroxidase–linked anti-mouse (#7076S) secondary reagent (Cell Signaling Technology).
Results
Loss of srp54 induces neutropenia in zebrafish
To understand the phenotype-genotype relationship in SRP54 deficiency, we characterized a novel zebrafish srp54 mutant (srp54sa11820), which carries a 3′ proximal adenosine-to-thymidine transversion that causes a premature stop codon in the N-domain of srp54, which effectively knocks out the gene function (Figure 1A; supplemental Figure 1A, available on the Blood Web site).18 Phenotypic analysis revealed that srp54sa11820/sa11820 (referred to as srp54−/−) embryos are developmentally impaired and show broad systemic defects such as the absence of blood flow, heart edema, reduced body size, pronounced body curvature, and other skeletal abnormalities (Figure 1B; supplemental Figure 1B). As a result of these developmental defects, srp54−/− zebrafish die at early embryonic stages, with first deaths being observed before 60 hpf and no embryo surviving after 72 hpf (Figure 1C). Conversely, srp54+/sa11820 (referred to as srp54+/−) zebrafish are viable and fertile and do not show any profound developmental defects (Figure 1B-C), indicating that the residual WT srp54 allele is sufficient to sustain tissue development. Of note, srp54−/− fish are incapable of breaking the chorion, which is in line with the assumption that protein secretion is impaired upon srp54 deficiency and might play a central role in the phenotypic manifestation (Figure 1D). However, WISH of embryos at 48 hpf using neutrophil-specific probes against myeloperoxidase (mpo) and lysozyme C (lyz) indicated reduced neutrophil numbers in both srp54−/− and srp54+/− embryos compared with WT embryos, with the effect being less severe in srp54+/− embryos (Figure 1E-F). These findings were confirmed by flow cytometric analyses using neutrophil-specific transgenic lines (supplemental Figure 1C).
![srp54+/− and srp54−/− fish display neutropenia but no overt pancreatic defects. (A) Structure of the human SRP54 NG domain. The cartoon shows the domain organization of SRP54, the sites of CN-relevant single-acid mutations (T115A, T117Δ, G226E [encircled]) and the truncation product of the srp54sa11820 variant in our zebrafish mutant (N-terminal 14 residues in blue). A nonhydrolyzable GTP-analog (GNP) taken from the SRP54/SRα structure is shown superposed (sticks) in the active site of the SRP54 G domain. (B) Representative images of srp54+/−, srp54−/−, and WT siblings. Fish were mounted in methylcellulose, and photographs were taken with a Zeiss SteREO Discovery.V20 microscope. (C) Kaplan-Meier survival analysis of genotyped fish (at least 45 embryos per condition from 3 biological replicates). (D) Representative images of WT and srp54−/− embryos after 54 hpf. Note that srp54−/− embryos are still inside the chorion. (E-F) Assessment of neutrophil counts in genotyped srp54+/−, srp54−/−, and WT siblings using WISH for (E) mpo or (F) lyz. Representative images are shown. Numbers below the representative images indicate the sum of embryos with the respective phenotype per total number of embryos analyzed in all replication experiments for the respective condition. (↓) Indicates downregulation. Shown are data from 3 biological replicates with at least 3 to 10 larvae per replicate. (G) Representative images after WISH for trypsin in srp54+/− and WT siblings at 96 hpf (left) and corresponding tissue slices (right). Student t test was used for statistical analysis. **P < .01; ***P < .005; ****P < .0001. Horizontal lines in the graphs represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/10/10.1182_blood.2020008115/2/m_bloodbld2020008115f1.png?Expires=1768789872&Signature=GN3Oe8bZshdzN-xpK2ENq91Ya3H2ZW3DKWSF4BGFaMx7jabqMl08hNnidHpe-BO0ieu~lVIE7XTstSvGNu0W0KN5sLY~Fx76CmA5z1bAIpCW2nx0n1vqY1HqBryEHUzbl6aOizuWk331RP7P7Wuf-R1aToXohGNDTNgObylXxRGMgkeeABSSJa-r1Nq1R9GTrZ~Z7zhfcKSOeS8dpEvbFMNdFI3j40tB45m34WeOA4xslhZEA64ndYy45TyIbN6eIIxeTTn6Iuaor80jsp0B9ax35Y6--VqyTBkjYBRxJ3tIrj6LYuc~fg6qgPqsAbEfef~5Owqk6RQsCLwPTj9tVA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
srp54+/− and srp54−/− fish display neutropenia but no overt pancreatic defects. (A) Structure of the human SRP54 NG domain. The cartoon shows the domain organization of SRP54, the sites of CN-relevant single-acid mutations (T115A, T117Δ, G226E [encircled]) and the truncation product of the srp54sa11820 variant in our zebrafish mutant (N-terminal 14 residues in blue). A nonhydrolyzable GTP-analog (GNP) taken from the SRP54/SRα structure is shown superposed (sticks) in the active site of the SRP54 G domain. (B) Representative images of srp54+/−, srp54−/−, and WT siblings. Fish were mounted in methylcellulose, and photographs were taken with a Zeiss SteREO Discovery.V20 microscope. (C) Kaplan-Meier survival analysis of genotyped fish (at least 45 embryos per condition from 3 biological replicates). (D) Representative images of WT and srp54−/− embryos after 54 hpf. Note that srp54−/− embryos are still inside the chorion. (E-F) Assessment of neutrophil counts in genotyped srp54+/−, srp54−/−, and WT siblings using WISH for (E) mpo or (F) lyz. Representative images are shown. Numbers below the representative images indicate the sum of embryos with the respective phenotype per total number of embryos analyzed in all replication experiments for the respective condition. (↓) Indicates downregulation. Shown are data from 3 biological replicates with at least 3 to 10 larvae per replicate. (G) Representative images after WISH for trypsin in srp54+/− and WT siblings at 96 hpf (left) and corresponding tissue slices (right). Student t test was used for statistical analysis. **P < .01; ***P < .005; ****P < .0001. Horizontal lines in the graphs represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
srp54+/− and srp54−/− fish display neutropenia but no overt pancreatic defects. (A) Structure of the human SRP54 NG domain. The cartoon shows the domain organization of SRP54, the sites of CN-relevant single-acid mutations (T115A, T117Δ, G226E [encircled]) and the truncation product of the srp54sa11820 variant in our zebrafish mutant (N-terminal 14 residues in blue). A nonhydrolyzable GTP-analog (GNP) taken from the SRP54/SRα structure is shown superposed (sticks) in the active site of the SRP54 G domain. (B) Representative images of srp54+/−, srp54−/−, and WT siblings. Fish were mounted in methylcellulose, and photographs were taken with a Zeiss SteREO Discovery.V20 microscope. (C) Kaplan-Meier survival analysis of genotyped fish (at least 45 embryos per condition from 3 biological replicates). (D) Representative images of WT and srp54−/− embryos after 54 hpf. Note that srp54−/− embryos are still inside the chorion. (E-F) Assessment of neutrophil counts in genotyped srp54+/−, srp54−/−, and WT siblings using WISH for (E) mpo or (F) lyz. Representative images are shown. Numbers below the representative images indicate the sum of embryos with the respective phenotype per total number of embryos analyzed in all replication experiments for the respective condition. (↓) Indicates downregulation. Shown are data from 3 biological replicates with at least 3 to 10 larvae per replicate. (G) Representative images after WISH for trypsin in srp54+/− and WT siblings at 96 hpf (left) and corresponding tissue slices (right). Student t test was used for statistical analysis. **P < .01; ***P < .005; ****P < .0001. Horizontal lines in the graphs represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
Interestingly, flow cytometric analyses of WKM (supplemental Figure 1D-E) in 2-year-old adult fish did not show differences in neutrophil counts between srp54+/− and WT fish, temporarily restricting the neutropenia of srp54+/− fish to early embryogenesis. This is in line with data from patients with neutropenia, which indicates that neutrophil counts can improve with age.33
Assessment of the size of the exocrine pancreas as a second hallmark of SDS by WISH using trypsin–specific probes showed no differences between WT and srp54+/− fish (Figure 1G). Analyzing the exocrine pancreas of srp54−/− embryos was not feasible, because these fish die before 72 hpf, when the first assessment of pancreas development can be performed in zebrafish embryos. Of note, injection of human WT SRP54 mRNA was able to transiently rescue embryonic lethality and neutropenia in srp54−/− fish (supplemental Figure 1F-G).
To investigate whether blood cell lineages other than granulocytes are affected by srp54 KO, we performed WISH of WT, srp54+/−, and srp54−/− zebrafish embryos using globin, runx1/c-myb, or rag1 probes, specific for erythrocytes, hematopoietic stem and progenitor cells (HSPCs), and lymphocytes, respectively (supplemental Figure 2). WISH using globin-specific probes revealed no differences between WT and srp54+/− embryos. Conversely, in srp54−/− fish, no globin signal was detectable in the periphery, indicating the absence of blood circulation as a result of heart edema. In agreement with that, globin signals were accumulating in the zebrafish heart (supplemental Figure 2).
No significant differences between the assessed genotypes were shown when using runx1/c-myb- and rag1-specific probes (supplemental Figure 2). Of note, rag1 expression could not be assessed in srp54−/− embryos because T cells are not detectable in the thymus before srp54−/− fish start to die.
Overexpression of mutated SRP54 induces an SDS-like phenotype in zebrafish embryos
Because all SRP54 mutations in patients are heterozygous, we aimed to elucidate whether the defects are dominant negative or functional nulls. We injected capped human mRNA from healthy and mutated SRP54 into srp54+/− zebrafish zygotes. After 2 dpf and 3 dpf, respectively, we performed WISH using mpo- and trypsin-specific probes. Interestingly, injection of T115A-, T117Δ-, and G226E- mutated human SRP54 mRNA reduced the number of neutrophils and the size of the exocrine pancreas, suggesting potential dominant-negative effects. Of note, the impacts of T117Δ were less severe compared with those of T115A and G226E, matching the phenotypes observed in patients (Figure 2A-D).1

Injections of mutated mRNAs into srp54+/− embryos induce an SDS-like phenotype, but the residual neutrophils are sufficient to be adequately recruited to injury sites. (A-D) Injection of T115A, T117Δ, or G226E human mRNA into srp54+/− embryos. (A-B) Representative images with (C-D) corresponding quantifications after WISH for mpo (A,C) and trypsin (B,D). (E) Fluorescent confocal microscopy images 8 hours after tail fin injury. Left, bright field; middle, fluorescence; right, merge. Yellow rectangle indicates the analyzed tail region. (F-H) Quantification of neutrophil migration of WT siblings, WT siblings injected with human G226E mRNA, srp54+/−, and srp54+/− zebrafish injected with human G226E mRNA. (F) Total number of mpo+ cells, and (G) mpo+ cells at wound. (H) Fraction of cells migrating toward the injury site. Images were acquired with a Point Scanning Confocal Leica SP5-II-MATRIX microscope (original magnification, ×10). Neutrophils were automatically counted using ImageJ software (we used 3 biological replicates with at least 2 to 3 larvae per replicate). Student t test was used for statistical analysis. ns, not significant. *P < .05; **P < .01; ***P < .005. Bar plots and horizontal lines in the graph represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
Injections of mutated mRNAs into srp54+/− embryos induce an SDS-like phenotype, but the residual neutrophils are sufficient to be adequately recruited to injury sites. (A-D) Injection of T115A, T117Δ, or G226E human mRNA into srp54+/− embryos. (A-B) Representative images with (C-D) corresponding quantifications after WISH for mpo (A,C) and trypsin (B,D). (E) Fluorescent confocal microscopy images 8 hours after tail fin injury. Left, bright field; middle, fluorescence; right, merge. Yellow rectangle indicates the analyzed tail region. (F-H) Quantification of neutrophil migration of WT siblings, WT siblings injected with human G226E mRNA, srp54+/−, and srp54+/− zebrafish injected with human G226E mRNA. (F) Total number of mpo+ cells, and (G) mpo+ cells at wound. (H) Fraction of cells migrating toward the injury site. Images were acquired with a Point Scanning Confocal Leica SP5-II-MATRIX microscope (original magnification, ×10). Neutrophils were automatically counted using ImageJ software (we used 3 biological replicates with at least 2 to 3 larvae per replicate). Student t test was used for statistical analysis. ns, not significant. *P < .05; **P < .01; ***P < .005. Bar plots and horizontal lines in the graph represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
To further investigate the previously mentioned dominant-negative effects, we injected capped human mRNA of either WT SRP54 or mutated SRP54 into WT zebrafish zygotes and again performed WISH using mpo- and trypsin-specific probes at 2 dpf and 3 dpf, respectively. Consistently, overexpression of mutated human SRP54 mRNA induced SDS phenotypes in zebrafish embryos with T117Δ causing the least severe defects (supplemental Figure 3A-B). Of note, injection of WT SRP54 mRNA as control had no effect. To assess the effects of mutated SRP54 in a genetic null background with no healthy srp54 alleles, we injected the 3 different mutated human mRNAs into srp54−/− fish (supplemental Figure 4). T115A and G226E did not rescue the neutrophil counts, but a partial rescue was observed upon injection of T117Δ, indicating residual functionality of this particular mutated protein version. However, ectopically applied T117Δ srp54 exerts detrimental effects on the healthy Srp54 protein (Figure 2; supplemental Figure 3), indicating that this mutant variant also acts in a dominant-negative way (supplemental Figure 4).
To investigate whether neutrophils were also qualitatively impaired by SRP54 mutations, we performed neutrophil migration assays in srp54+/− zebrafish. After inducing a tail wound in 2-dpf zebrafish embryos followed by incubation for 8 hours, the total number of neutrophils and the number of neutrophils at the injury site were measured (Figure 2E-H). Total numbers of neutrophils showed the same trends as had been previously observed in noninjured embryos: srp54+/− displayed fewer neutrophils compared with WT embryos, and injection of G226E human mRNA further reduced the neutrophil counts in a dominant-negative manner (Figure 2F). The number of neutrophils at the injury site was not significantly altered between the different srp54 genotypes and also not upon G226E injection, which indicates persistent functional integrity of residual neutrophils (Figure 2G). Given the differences in total neutrophil counts and the constant number of neutrophils at the injury site, the relative proportion of cells at the injury site was increased in srp54+/− embryos or upon injection of G226E mRNA. In sum, these data suggest that neutrophil migration to an injury site is neither impaired in srp54+/− embryos alone nor in srp54+/− embryos injected with G226E mRNA and that the residual functional Srp54 protein in these fish is sufficient to sustain the migratory ability of neutrophils (Figure 2H).
Dominant-negative effects of SRP54 mutations are conserved in human cells
To explore the conservation of the dominant-negative effects of mutated SRP54 in human cells, we first lentivirally transduced the promyelocytic HL-60 cell line, known to differentiate upon ATRA treatment, with the 3 identified SRP54 mutations (T115A, T117Δ, and G226E; Figure 3A). Successful integration and expression of proteins was assessed by western blotting (Figure 3B). After treatment with ATRA for 6 days, the majority of cells showed neutrophil characteristics and expressed CD15 and CD16 (supplemental Figure 5A-B).34 To visualize potential impairment by the expression of mutant SRP54, we assessed nuclear lobulation as a feature of granulocytic differentiation using hematoxylin and eosin–stained cytospots (Figure 3A,C). Compared with WT transduced and empty control cells, the cells expressing mutated SRP54 alleles showed markedly reduced lobulation of neutrophilic nuclei (Figure 3D). In addition, the levels of CD11b (a surface marker of mature granulocytes) were significantly decreased upon T115A and G226E expression (Figure 3E-F). Of note, T117Δ affected granulocytic differentiation to a lesser degree than either T115A or G226E, again revealing the overall milder dominant-negative effects of T117Δ, which is consistent with its expression in CN rather than in SDS-like disease.
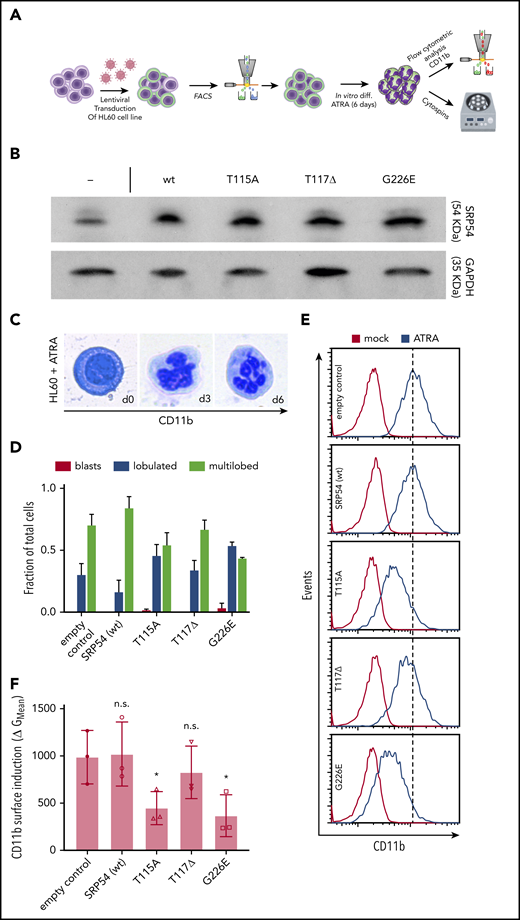
Dominant-negative effects of SRP54 mutations are conserved in the human HL-60 cell line and lead to differentiation defects. (A) Schematic overview of the experimental setup. (B) Western blots document elevated SRP54 protein expression (top) in HL-60 cells upon transduction with mutant or wt SRP54 constructs. Nontransduced control cells (–) are shown on the left. The following expression fold changes were deduced from area counts relative to empty control as quantified by Fiji software: wt, 2.8; T115A, 2.9; T117Δ, 1.9; and G226E, 3.5. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control and for normalization. (C) Representative images of cells during ATRA-driven HL-60 cell differentiation (left: blast cell, day 0; middle: lobulated neutrophil, day 3; right: multilobulated neutrophil, day 6). Photographs were taken from hematoxylin and eosin (H&E)–stained cytospots. (D) Quantification of H&E-stained cytospots after 6 days of treatment with ATRA. Criteria for classification: No lobules indicates blasts; 1 to 5 lobules indicates lobulated; >5 lobules indicates multilobed. Statistics: empty vs SRP54 (wt): blasts, lobulated, and multilobed are not significant; empty vs T115A: blasts are not significant and lobulated and multilobed have P < .05; empty vs T117Δ: blasts, lobulated, and multilobed are not significant; empty vs G226E: blasts are not significant and lobulated and multilobed have P < .005. (E) Histograms indicating CD11b surface staining on empty control, SRP54 (WT), T115A, T117Δ, and G226E transduced HL-60 cells as analyzed by flow cytometry. (F) Corresponding quantification of CD11b expression on empty control, SRP54 (WT), T115A, T117Δ, and G226E transduced cells. Plot shows the geometric (G) mean fluorescence intensity shift in the CD11b channel upon treatment with ATRA (y-axis) per indicated cell lines (x-axis). Note slightly reduced CD11b surface induction in T117Δ cells but a significant differentiation block in cells expressing T115A and G226E mutant forms of SRP54. Student t test was used for statistical analysis. diff, differentiation; FACS, fluorescence-activated cell sorting. *P < .05. Bar plots represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
Dominant-negative effects of SRP54 mutations are conserved in the human HL-60 cell line and lead to differentiation defects. (A) Schematic overview of the experimental setup. (B) Western blots document elevated SRP54 protein expression (top) in HL-60 cells upon transduction with mutant or wt SRP54 constructs. Nontransduced control cells (–) are shown on the left. The following expression fold changes were deduced from area counts relative to empty control as quantified by Fiji software: wt, 2.8; T115A, 2.9; T117Δ, 1.9; and G226E, 3.5. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control and for normalization. (C) Representative images of cells during ATRA-driven HL-60 cell differentiation (left: blast cell, day 0; middle: lobulated neutrophil, day 3; right: multilobulated neutrophil, day 6). Photographs were taken from hematoxylin and eosin (H&E)–stained cytospots. (D) Quantification of H&E-stained cytospots after 6 days of treatment with ATRA. Criteria for classification: No lobules indicates blasts; 1 to 5 lobules indicates lobulated; >5 lobules indicates multilobed. Statistics: empty vs SRP54 (wt): blasts, lobulated, and multilobed are not significant; empty vs T115A: blasts are not significant and lobulated and multilobed have P < .05; empty vs T117Δ: blasts, lobulated, and multilobed are not significant; empty vs G226E: blasts are not significant and lobulated and multilobed have P < .005. (E) Histograms indicating CD11b surface staining on empty control, SRP54 (WT), T115A, T117Δ, and G226E transduced HL-60 cells as analyzed by flow cytometry. (F) Corresponding quantification of CD11b expression on empty control, SRP54 (WT), T115A, T117Δ, and G226E transduced cells. Plot shows the geometric (G) mean fluorescence intensity shift in the CD11b channel upon treatment with ATRA (y-axis) per indicated cell lines (x-axis). Note slightly reduced CD11b surface induction in T117Δ cells but a significant differentiation block in cells expressing T115A and G226E mutant forms of SRP54. Student t test was used for statistical analysis. diff, differentiation; FACS, fluorescence-activated cell sorting. *P < .05. Bar plots represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
SRP54 mutant alleles impair granulocytic differentiation of CD34+ cord blood cells
Importantly, similar results were observed with transduced healthy hematopoietic cord blood–derived CD34+ cells, in which flow cytometric analyses of CD11b surface levels of CD15 and CD16 double-positive cells revealed that exogenous expression of p.G226E significantly impaired in vitro differentiation toward neutrophil fate compared with WT SRP54 transduced cells (Figure 4A-D; supplemental Figure 6A-B). Of note, the exogenous expression of p.G226E SRP54, but not p.T115A or p.T117Δ, was investigated in CD34+ HSPCs because the most profound phenotypes were expected with this mutation, according to previous findings and patient data.
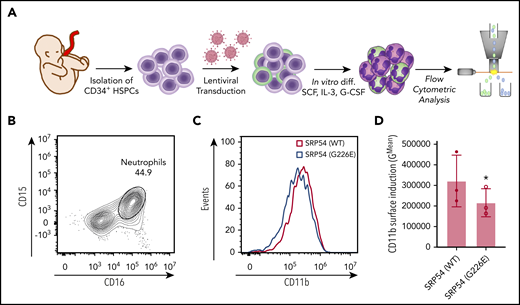
SRP54-mutant alleles impair granulocytic differentiation of CD34+ cord blood cells. (A) Schematic overview of the experimental setup for isolation, lentiviral transduction, and in vitro cultivation of CD34+ cord blood cells followed by flow cytometric analyses. (B) Gating strategy to identify CD15 and CD16 double-positive neutrophils. (C) Histograms of flow cytometric analyses of CD11b levels of SRP54 transduced and differentiated CD34+ cells. (D) Flow cytometric quantification of CD11b expression of transduced and differentiated CD34+ cells. Plot shows the geometric mean fluorescence intensity of WT and G226E transduced cells. A ratio paired Student t test was used for statistical analysis. G-CSF, granulocyte colony-stimulating factor; IL-3, interleukin 3; SCF, stem cell factor. *P < .05. Bar plots represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
SRP54-mutant alleles impair granulocytic differentiation of CD34+ cord blood cells. (A) Schematic overview of the experimental setup for isolation, lentiviral transduction, and in vitro cultivation of CD34+ cord blood cells followed by flow cytometric analyses. (B) Gating strategy to identify CD15 and CD16 double-positive neutrophils. (C) Histograms of flow cytometric analyses of CD11b levels of SRP54 transduced and differentiated CD34+ cells. (D) Flow cytometric quantification of CD11b expression of transduced and differentiated CD34+ cells. Plot shows the geometric mean fluorescence intensity of WT and G226E transduced cells. A ratio paired Student t test was used for statistical analysis. G-CSF, granulocyte colony-stimulating factor; IL-3, interleukin 3; SCF, stem cell factor. *P < .05. Bar plots represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
Insufficient xbp1 splicing drives the SDS-like phenotype in srp54-defective zebrafish embryos
XBP1 is one of the key transcription factors involved in the unfolded protein response (UPR).35-37 However, it functions as an active transcription factor only if its mRNA is cleaved by the transmembrane endoribonuclease IRE1 in a process termed unconventional splicing. In conditions of ER stress, unconventional splicing of XBP1 predominantly takes place at the ER membrane.38,39 The unspliced mRNA of XBP1 (XBP1u) is thereby transported to the ER membrane in a complex together with the ribosome and its own nascent polypeptide chain in an SRP-dependent manner (Figure 5A).40 This dependence on a functional SRP and its previously established importance for neutrophil differentiation41 caused us to hypothesize that the splicing of XBP1 mRNA might be a key factor contributing to the phenotypic manifestation of SRP54 mutations in patients.
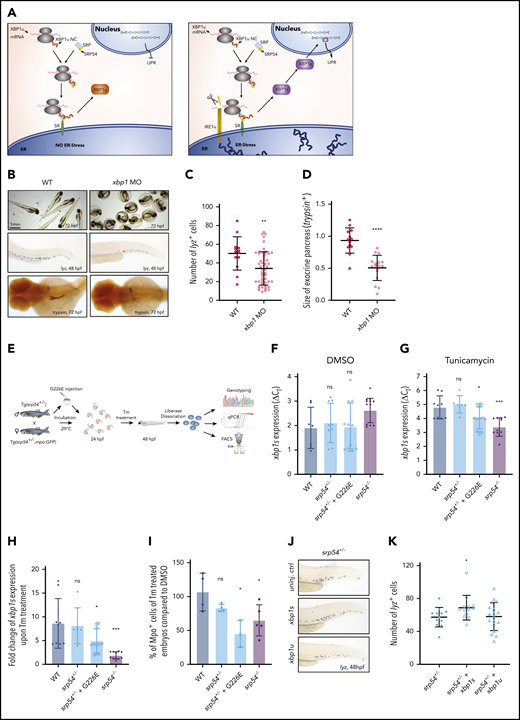
Insufficient xbp1 splicing drives the SDS phenotype in srp54-defective zebrafish embryos. (A) Graphical representation of the unconventional splicing of XBP1. Left: cell without or with moderate ER stress; right: cell under ER stress conditions. (B) Representative images of WT embryos compared with xbp1 morphants. Top row: photos showing that xbp1 morphants are incapable of hatching and breaking the chorion at 72 hpf; middle row: WISH using lyz-specific probes; bottom row: WISH using trypsin-specific probes. (C) Quantification of neutrophils using lyz-specific probes. (D) Measurement of the exocrine pancreas using trypsin-specific probes. The size of the exocrine pancreas was semi-automatically measured using ImageJ software. (E) Schematic overview of the experimental setup to assess xbp1 levels in zebrafish embryos. (F) Differences of the cycle threshold (CT) between xbp1s and the housekeeping gene gapdh (ΔCT values) of dissolved cells from dimethyl sulfoxide (DMSO)–treated WT, srp54+/−, srp54+/− injected with human G226E mRNA, and srp54−/− embryos measured by qRT-PCR. (G) ΔCT values for xbp1s of dissolved cells from Tm-treated WT, srp54+/−, srp54+/− injected with human G226E mRNA and srp54−/− embryos measured by qRT-PCR. (H) Fold change of xbp1s expression upon Tm treatment compared with DMSO treatment: WT, srp54+/−, srp54+/− injected with human G226E mRNA and srp54−/− embryos (we used 3 biological replicates with at least 2 larvae per replicate). (I) Percentage of Mpo+ cells from Tm-treated compared with DMSO-treated embryos measured by flow cytometry. Tg(srp54+/−;mpo:eGFP) zebrafish were incrossed, and their progeny were genotyped and analyzed by flow cytometry. A minimum of 5 embryos were pooled and dissociated per biological replicate. Each dot represents a biological replicate. WT, srp54+/−, srp54+/− injected with human G226E mRNA and srp54−/− embryos. (J) Representative images of WISH using lyz-specific probes performed on srp54+/− embryos either uninjected (top), injected with xbp1s mRNA (middle), or injected with xbp1u mRNA (bottom). (K) Quantification of the number of lyz+ cells (we used 4 biological replicates with at least 3 larvae per replicate). Student t test was used for statistical analysis: SR, signal receptor. *P < .05; **P < .01; ***P < .005; ****P < .0001. Bar plots and horizontal lines in the graphs represent the mean value of the replicates. Error bars indicate the standard deviation of the mean. NC, nascent chain.
Insufficient xbp1 splicing drives the SDS phenotype in srp54-defective zebrafish embryos. (A) Graphical representation of the unconventional splicing of XBP1. Left: cell without or with moderate ER stress; right: cell under ER stress conditions. (B) Representative images of WT embryos compared with xbp1 morphants. Top row: photos showing that xbp1 morphants are incapable of hatching and breaking the chorion at 72 hpf; middle row: WISH using lyz-specific probes; bottom row: WISH using trypsin-specific probes. (C) Quantification of neutrophils using lyz-specific probes. (D) Measurement of the exocrine pancreas using trypsin-specific probes. The size of the exocrine pancreas was semi-automatically measured using ImageJ software. (E) Schematic overview of the experimental setup to assess xbp1 levels in zebrafish embryos. (F) Differences of the cycle threshold (CT) between xbp1s and the housekeeping gene gapdh (ΔCT values) of dissolved cells from dimethyl sulfoxide (DMSO)–treated WT, srp54+/−, srp54+/− injected with human G226E mRNA, and srp54−/− embryos measured by qRT-PCR. (G) ΔCT values for xbp1s of dissolved cells from Tm-treated WT, srp54+/−, srp54+/− injected with human G226E mRNA and srp54−/− embryos measured by qRT-PCR. (H) Fold change of xbp1s expression upon Tm treatment compared with DMSO treatment: WT, srp54+/−, srp54+/− injected with human G226E mRNA and srp54−/− embryos (we used 3 biological replicates with at least 2 larvae per replicate). (I) Percentage of Mpo+ cells from Tm-treated compared with DMSO-treated embryos measured by flow cytometry. Tg(srp54+/−;mpo:eGFP) zebrafish were incrossed, and their progeny were genotyped and analyzed by flow cytometry. A minimum of 5 embryos were pooled and dissociated per biological replicate. Each dot represents a biological replicate. WT, srp54+/−, srp54+/− injected with human G226E mRNA and srp54−/− embryos. (J) Representative images of WISH using lyz-specific probes performed on srp54+/− embryos either uninjected (top), injected with xbp1s mRNA (middle), or injected with xbp1u mRNA (bottom). (K) Quantification of the number of lyz+ cells (we used 4 biological replicates with at least 3 larvae per replicate). Student t test was used for statistical analysis: SR, signal receptor. *P < .05; **P < .01; ***P < .005; ****P < .0001. Bar plots and horizontal lines in the graphs represent the mean value of the replicates. Error bars indicate the standard deviation of the mean. NC, nascent chain.
To verify our hypothesis, we knocked down xbp1 in zebrafish embryos by MO injection (supplemental Figure 7A-B). Notably, like srp54−/− embryos, xbp1 morphants were incapable of breaking the chorion (Figure 5B; Bennet et al42 ). Furthermore, WISH using lyz- and trypsin-specific probes revealed a significant reduction in the number of neutrophils and of the size of the exocrine pancreas (Figure 5B-D), whereas WISH using globin-, runx1/c-myb–, or rag1-specific probes did not show any additional hematopoietic impairment (supplemental Figure 7C-D).
In the next step, we analyzed the levels of spliced xbp1 (xbp1s) in uninjected WT, srp54+/−, and srp54−/− zebrafish embryos and upon injection of G226E-mutated human SRP54 mRNA into srp54+/− embryos (Figure 5E). qRT-PCR of total body cells revealed that srp54−/− embryos showed increased xbp1s mRNA levels, indicating that the splicing of xbp1 is not completely abolished upon homozygous srp54 KO (Figure 5F). To determine whether xbp1 splicing is impaired in srp54-defective zebrafish, we experimentally induced ER stress in all genotypes by treating the embryos with Tm (Figure 5E). Interestingly, after Tm treatment, srp54−/− and srp54+/− zebrafish injected with G226E mRNA showed reduced levels of xbp1s compared with WT and srp54+/− zebrafish (Figure 5G-H). These effects were specific for xbp1s, since the expression of other UPR players such as atf4, bip, and chop was unaffected by srp54 KO (supplemental Figure 7E).
To analyze whether the impaired splicing capability of srp54−/− and G226E-injected srp54+/− fish induced neutropenia, we used flow cytometry to evaluate the number of neutrophils in embryos treated with dimethyl sulfoxide (DMSO) and Tm. Strikingly, Tm treatment significantly lowered the number of Mpo+ cells in srp54−/− fish and in srp54+/− fish injected with G226E mRNA (Figure 5I) compared with DMSO treatment.
Finally, to functionally explore the relevance of impaired xbp1 splicing for the phenotypes associated with srp54 deficiency, we injected zebrafish mRNA from xbp1s and xbp1u into srp54+/− embryos. Indeed, only xbp1s mRNA, but not xbp1u mRNA, was able to significantly rescue the neutrophil numbers in srp54-deficient embryos (Figure 5J-K).
Impaired XBP1 splicing is conserved in human SRP54-mutant cells
To uncover potential conservation of the impairment of XBP1 splicing in human cells, we treated transduced human HL-60 cells expressing either WT or G226E SRP54 with Tm and measured the levels of XBP1s. Importantly, G226E SRP54-expressing cells showed significantly lower levels of XBP1s after treatment with Tm compared with WT transduced cells and presented a more than fivefold decreased capability to splice XBP1u under ER stress conditions, thereby matching our findings in zebrafish srp54 mutants (Figure 6A-C).

XBP1 splicing defects are conserved in human HL-60 cells with mutant SRP54 expression. (A) ΔCT values for XBP1s expression of DMSO-treated SRP54 (WT) transduced HL-60 cells compared with SRP54 (G226E) transduced HL-60 cells measured by qRT-PCR. (B) ΔCT values for XBP1s for Tm-treated SRP54 (WT) compared with SRP54 (G226E) transduced HL-60 cells measured by qRT-PCR. (C) Fold change of XBP1s expression upon Tm treatment compared with DMSO treatment of SRP54 (WT) and SRP54 (G226E) transduced HL-60 cells. We used 3 biological replicates. Student t test was used for statistical analysis. ***P < .005; ****P < .0001. Bar plots represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
XBP1 splicing defects are conserved in human HL-60 cells with mutant SRP54 expression. (A) ΔCT values for XBP1s expression of DMSO-treated SRP54 (WT) transduced HL-60 cells compared with SRP54 (G226E) transduced HL-60 cells measured by qRT-PCR. (B) ΔCT values for XBP1s for Tm-treated SRP54 (WT) compared with SRP54 (G226E) transduced HL-60 cells measured by qRT-PCR. (C) Fold change of XBP1s expression upon Tm treatment compared with DMSO treatment of SRP54 (WT) and SRP54 (G226E) transduced HL-60 cells. We used 3 biological replicates. Student t test was used for statistical analysis. ***P < .005; ****P < .0001. Bar plots represent the mean value of the replicates. Error bars indicate the standard deviation of the mean.
Discussion
Our data in homozygous srp54 KO zebrafish embryos indicate that the Srp54 protein is critically required for the development of multiple tissues with neutrophils as cells with the highest dependency on proper SRP54 levels. The fact that complete loss of srp54 is embryonically lethal might be the reason why no homozygous SRP54 defects have yet been identified in patients.1,14 Interestingly, srp54+/− zebrafish are viable, healthy, and fertile, and they suffer from only a mild form of neutropenia. Because all known patients with SRP54-associated neutropenia carry heterozygous defects but still show variable and also more severe clinical phenotypes, we hypothesized that the underlying mutations must have dominant-negative effects that compete with, and may effectively override, the endogenous SRP54 functions derived from the persisting WT allele. Exogenous expression of mutated human SRP54 in srp54+/− or WT zebrafish embryos as well as in human HL-60 cells or CD34+ HSPCs confirmed these dominant-negative effects. In detail, HL-60 cells and healthy cord blood–derived CD34+ HSPCs showed impaired granulocytic differentiation upon transduction with mutated human SRP54, but not upon transduction with WT human SRP54, indicating that SRP54 overexpression is not disease inducing per se but that observed phenotypes are specifically mediated by pathologic effects of mutated SRP54.
Importantly, the granulocytic differentiation defects observed in HL-60 cells and CD34+ HSPCs represent a novel cellular process, which, in addition to proliferative defects described elsewhere, ER stress, apoptosis, and autophagy, contributes to the neutropenic phenotype associated with SRP54 deficiency.14
Of note, the migratory function of neutrophils in zebrafish embryos was not altered upon heterozygous srp54 KO and G226E expression. The absolute number of neutrophils at the injury site was constant among the different conditions, indicating that the residual functional Srp54 protein in the investigated embryos was sufficient to sustain the migration capacity of mutant neutrophils. This finding adds a novel perspective to previous results shown by Carapito et al,1 in which MO-driven knockdown of srp54 led to neutrophil migratory defects. Considering that srp54 morphants had lower levels of functional Srp54 protein compared with the srp54+/− or G226E-injected fish analyzed herein (also noticeable by the severe form of neutropenia and strong exocrine pancreatic defects compared with only a mild form of neutropenia and absent or weak exocrine pancreatic defects, respectively; supplemental Figure 1A), we can conclude that a certain level of WT Srp54 protein needs to be present to sustain the neutrophil migratory function. Furthermore, the unaffected migratory ability of the neutrophils in the zebrafish model we investigated indicates that increased infection rates observed in patients are probably a result of the overall reduced numbers of neutrophils or other potentially affected functions such as the formation of neutrophil extracellular traps or the release of antimicrobial enzymes.
To investigate why and how srp54 defects specifically impair neutrophil differentiation, we aimed to understand the underlying mechanistic details. As shown before,14 SRP54 mutations lead to ER stress. The ER stress conditions might be the reason why neutrophils but no other blood cells are affected, since Tanimura et al revealed that ER stress needs to be absent and UPR needs to be active to allow neutrophil, but not macrophage, differentiation.41 However, in the case of SRP54-mediated CN, it is still unclear how elevated ER stress develops. We hypothesized that the important UPR mediator XBP1 might be contributing to the disease manifestation, because its activation by unconventional splicing is dependent on the SRP, and SRP54 knockdown in HELA cells was associated with dampened XBP1 splicing.40 Of note, Bellanné-Chantelot et al14 showed that the XBP1s expression is elevated in SRP54-deficient patient cells compared with healthy control cells. However, assuming that ER stress was present in patient-derived cells, genes involved in UPR would naturally be upregulated. To allow a comparison regarding xbp1 splicing, we sought to induce ER stress in control cells. We treated zebrafish embryos as well as human HL-60 cells with Tm, a natural ER stress inducer. Here we show that insufficient splicing of XBP1 contributes to elevated ER stress levels in SRP54-defective human cells and zebrafish and that knockdown of xbp1 in zebrafish embryos phenocopies several characteristics of srp54−/− mutants. Furthermore, exogenous expression of xbp1s in srp54+/− zebrafish was able to rescue the neutropenic phenotype, directly linking impaired xbp1 splicing to disease manifestation and advocating the IRE1-XBP1 axis as a potential therapeutic target of SRP54 deficiencies.
In Figure 7, we summarize our findings and provide a hypothetical model explaining how SRP54 mutations may impair XBP1 splicing. In this model, the binding of the SRP to the signal receptor (SR) is destabilized by mutations in SRP54. Consequently, XBP1u mRNA does not get close to the ER membrane resident endonuclease IRE1 and can no longer be spliced, which leads to the absence of the UPR mediator XBP1s, which eventually leads to unresolved ER stress. The dominant-negative effects of SRP54 mutations are thus a result of the functionally impaired mutant SRP54 protein competing with endogenous WT SRP54 for signal peptides of proteins that need to be secreted. Consequently, the nascent chain-ribosome complexes of these proteins are no longer available for the functional WT SRP54 and cannot be transported to the ER membrane.
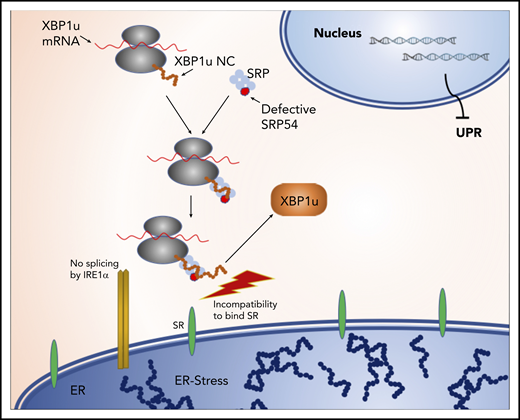
Model of impaired XBP1 splicing upon SRP54 mutations. The binding of the SRP to the SR is destabilized by mutations in SRP54. Consequently, XBP1u mRNA does not get spliced by IRE1, leading to the absence of the UPR mediator XBP1s, which eventually leads to unresolved ER stress.
Model of impaired XBP1 splicing upon SRP54 mutations. The binding of the SRP to the SR is destabilized by mutations in SRP54. Consequently, XBP1u mRNA does not get spliced by IRE1, leading to the absence of the UPR mediator XBP1s, which eventually leads to unresolved ER stress.
Our proposed model is in agreement with the findings of Goldberg et al,43 in which the deletion of threonine115 in SRP54 leads to the destabilization of the SRP-SR complex.
Taken together, we have provided a stable in vivo model for studies of SRP54, which reveal novel mechanistic insights into the function of SRP54 and associated mutations in the regulation of ER stress response and neutrophil development. Given the ubiquitous expression of SRP54 and its fundamental requirement for various developmental processes, dose-dependent SRP54 effects likely play important roles in the pathophysiology of other tissues as well.
For original data, please send an e-mail to Martina Konantz at martina.konantz@unibas.ch or Claudia Lengerke at claudia.lengerke@med.uni-tuebingen.de.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the members of the Imaging Core Facility of the Biozentrum, University of Basel (Basel, Switzerland) and the DBM Microscopy Facility (Basel, Switzerland) for assistance with imaging and the members of the DBM Flow Cytometry Facility (Basel, Switzerland) for assistance with flow cytometry.
This work was supported by grants from the Swiss National Science Foundation (149735) (C.L.) and the Interreg V European Regional Development Fund (European Union) program (project 3.2 TRIDIAG) (R.C., S.B., and C.L.). The laboratory of S.B. is funded by grants from the Agence Nationale de la Recherche (ANR) (ANR-11-LABX-0070_TRANSPLANTEX) and MSD-Avenir grant (AUTOGEN).
Authorship
Contribution: C.S., M.K., and C.L. designed the study, analyzed the data, and wrote the manuscript; C.S., M.K., P.H., J.S.M., and J. Schärer performed zebrafish experiments and interpreted data; C.S., A.D., M.A., and T.S. performed in vitro experiments and interpreted data; I.S. and K. Wild performed in silico structure function mapping; R.C., S.B., K. Welte, and J. Skokowa interpreted data; and all authors contributed to writing and approving the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Claudia Lengerke, University Hospital Tübingen, Otfried-Müller-Str 10, Tuebingen, 72076 Germany; e-mail: claudia.lengerke@med.uni-tuebingen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal