

Key Points
We report autosomal-recessive germline deficiency of the methylcytosine dioxygenase TET2 in 3 immunodeficient children.
Their phenotype of immunodeficiency, autoimmunity and lymphoproliferation highlights requisite roles for TET2 in the human immune system.

Abstract
Molecular dissection of inborn errors of immunity can help to elucidate the nonredundant functions of individual genes. We studied 3 children with an immune dysregulation syndrome of susceptibility to infection, lymphadenopathy, hepatosplenomegaly, developmental delay, autoimmunity, and lymphoma of B-cell (n = 2) or T-cell (n = 1) origin. All 3 showed early autologous T-cell reconstitution following allogeneic hematopoietic stem cell transplantation. By whole-exome sequencing, we identified rare homozygous germline missense or nonsense variants in a known epigenetic regulator of gene expression: ten-eleven translocation methylcytosine dioxygenase 2 (TET2). Mutated TET2 protein was absent or enzymatically defective for 5-hydroxymethylating activity, resulting in whole-blood DNA hypermethylation. Circulating T cells showed an abnormal immunophenotype including expanded double-negative, but depleted follicular helper, T-cell compartments and impaired Fas-dependent apoptosis in 2 of 3 patients. Moreover, TET2-deficient B cells showed defective class-switch recombination. The hematopoietic potential of patient-derived induced pluripotent stem cells was skewed toward the myeloid lineage. These are the first reported cases of autosomal-recessive germline TET2 deficiency in humans, causing clinically significant immunodeficiency and an autoimmune lymphoproliferative syndrome with marked predisposition to lymphoma. This disease phenotype demonstrates the broad role of TET2 within the human immune system.
Introduction
Inborn errors of immunity (IEIs) are rare inherited diseases caused by aberrations in the genome,1,2 leading to functional abnormalities in the immune system that range from susceptibility to infection to immune dysregulation. Predisposition to blood cancers is a relatively frequent manifestation of IEI; in many cases, it is directly attributable to the effect of the disease-causing mutation on lymphocyte behavior.3 One such example is provided by autoimmune lymphoproliferative syndrome (ALPS), in which uncontrolled nonmalignant lymphoproliferation results from impaired apoptosis of lymphocytes and results in a high risk for development of lymphomas and autoimmune phenomena, such as cytopenias.4-7 Not surprisingly, there is an overlap between the genes that harbor germline mutations causing immune dysregulation disorders and genes that commonly acquire somatic variants in the context of sporadic blood cancers.
Here, we report for the first time 2 novel germline loss-of-function (LOF) point mutations in ten-eleven translocation methylcytosine dioxygenase 2 (TET2), causing an autosomal-recessive syndrome of immunodeficiency with lymphoproliferative disease in 3 children from 2 unrelated consanguineous families. Consistent with a tumor suppressor role, somatic TET2 LOF mutations are frequently observed in hematopoietic disorders, myeloid and lymphoid malignancies, and clonal hematopoiesis of indeterminate potential (CHIP).8-14 TET2, along with TET1 and TET3, is a member of the TET family of epigenetic regulators, whose enzymatic function is to convert 5-methylcytosine (5mC), an important epigenetic modification of the mammalian genome, to 5-hydroxymethylcytosine (5hmC) and additional oxidation products in a pathway of active DNA demethylation.15,16 Furthermore, TETs interact with histone-modifying enzymes and transcription factors to exert additional epigenetic effects. Thus, TET2 activity shapes the local chromatin environment, especially at active enhancers and cell type–specific regulatory regions, influencing their accessibility to master transcription factors.17-19
TET2 is highly expressed in hematopoietic stem/progenitor cells and plays important roles in hematopoiesis, including regulating the self-renewal of stem cells, lineage commitment, and differentiation of monocytes.20-22 Effects on the transcriptional program and function of lymphocytes have been proposed in several murine models of TET2 deficiency that are viable, fertile, and develop normally but demonstrate myeloproliferation, splenomegaly, monocytosis, myeloproliferative diseases, and lymphomagenesis.23-28
In this study, we define the molecular and clinical consequences of homozygous germline TET2 impairment in 3 immunodeficient children that lead to altered DNA methylation and B-cell maturation, skewed T-cell differentiation and hematopoiesis, and development of lymphomas in childhood.
Methods
Detailed protocols, including antibodies used, are provided as supplemental Material, available on the Blood Web site.
Patient cohort
The parents of all patients and healthy donor volunteers provided written informed consent to participate in research protocols approved by the local Research Ethics Committee.
Genetic analysis
Patient genomic DNA was subjected to whole-exome sequencing (WES) and analyzed by standard methods. This revealed candidate disease-causing variants in TET2, which were confirmed by Sanger sequencing. The patients’ lymphoma tumor samples were analyzed for additional somatic mutations by WES.
TET2 protein expression and enzyme activity
Expression of TET2 protein was determined by immunoblotting. Enzymatic activity was assessed by immunofluorescence staining of 5hmC after transfection of the HEK293T cell line with wild-type (wt) or mutated TET2H1382R.
DNA methylation assay and 5hmC staining
Methylation and 5-hydroxymethylation of DNA were analyzed by enzymatic assay, involving glucosylation followed by enzyme restriction with MspI (R0106S) and HpaII (R0171S) at the DNA sequence 5′-CCGG-3′. MspI cleaves 5mC and 5hmC, but not glucosylated 5hmC, whereas any modification with 5mC, 5hmC, or glucosylated 5hmC at either cytosine will prevent cleavage by HpaII.
B-cell differentiation assay
Isolated peripheral blood B cells were stimulated with CD40L and F(ab′)2 anti-immunoglobulin G (IgG)/IgM, mimicking a T-cell–dependent immune response. The immunophenotype of differentiating cells was analyzed by flow cytometry, and the secretion of IgG and IgM was detected in parallel by enzyme-linked immunosorbent assay.
iPSC derivation and hematopoietic differentiation
Patients’ fibroblasts were reprogrammed into induced pluripotent stem cells (iPSCs) using Yamanaka factors, characterized for their pluripotent potential, and differentiated to hematopoietic precursors in vitro. The proportion of erythro-megakaryocytic and myeloid progenitors in culture and their clonogenic potential were determined by flow cytometry and colony-forming unit (CFU) assay, respectively.
FasL-induced apoptosis assay by annexin V/propidium iodide staining
Peripheral blood mononuclear cells (PBMCs), blasted with phytohemagglutinin and interleukin-2 (IL-2), were stimulated with a soluble Fas ligand (FasL) set, stained with Annexin V-FITC and propidium iodide, and analyzed by flow cytometry.
Statistical analysis
Data are mean ± standard deviation (SD) of ≥2 independent experiments. Nonparametric 1-way analysis of variance Kruskal-Wallis with Dunn’s multiple-comparisons test, 2-tailed Student t test, and unpaired Student t test were used to calculate statistical significance (P < .05) using GraphPad Prism 7.02 software.
Results
Case descriptions
Patient 1 (P1) was the second-born child of related parents. He presented to the hospital at 4 weeks of age with lower respiratory tract infection (LRTI), associated with respiratory syncytial virus and cytomegalovirus, and was treated with ganciclovir. Subsequently, P1 showed failure to thrive and developmental delay and was hospitalized frequently for LRTIs. From 18 months of age, he developed autoimmune cytopenias requiring frequent transfusions, accompanied by hepatosplenomegaly, chronic lymphadenopathy, hypergammaglobulinemia, and persistent Epstein-Barr virus (EBV) viremia. The diagnosis of ALPS-U was confirmed with the demonstration of defective Fas-mediated apoptosis (Figure 1A) and increased double-negative (CD4−CD8−) TCRαβ T cells (DNTs; 20%) (Table 1) in the absence of known genetic causes of ALPS.29
![Patients’ clinical and laboratory characteristics. (A) FasL-induced apoptosis in patients’ and healthy controls’ phytohemagglutinin- and IL-2–stimulated T-blasts determined by flow cytometry using annexin V/propidium iodide staining showed impaired apoptosis in P1 and P2 before transplantation, normal response in P3 before transplantation, and repaired response in P1 after transplantation compared with healthy control cells. (B) Histopathology of all 3 patients’ lymphoid tumors. P1: EBV+ Hodgkin-like polymorphic B-cell lymphoproliferative disorder; P2: nodal peripheral T-cell lymphoma with follicular helper T-cell phenotype; P3: primary mediastinal large B-cell lymphoma (upper panels [original magnification ×600]: hematoxylin and eosin staining; lower panels [original magnification ×400]: EBV, Epstein-Barr virus encoded RNA in situ hybridization, and CD3 and CD20 immunohistochemical staining). (C) Rapid autologous lymphocyte reconstitution after HSCT in all 3 patients with homozygous TET2 LOF, despite full T-cell–depleting serotherapy. Asterisks indicate the first measurement of T-cell chimerism in each patient: P1 (day +28) 78% recipient; P2 (day +46), 100% recipient; P3 (day +52), 91% recipient.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/9/10.1182_blood.2020005844/3/m_bloodbld2020005844f1.png?Expires=1769090859&Signature=TQwfQ7T0hRU6hZKwen4JnuBc6~xD6WOSG8QgayU0UPep8qOf6t6Nm3Y4LFqChcRfntGMIhwkbymjY8WUPua5u5iUeOmimuwAuUIrcn1kM5iZAf~IkLaBcAHR0eHXSPtVDWFcCaOqgoO6kMEWtcS5zbTbLxqjrffvRX8kkifqlxYznWZvFiKpLnl4yNZPdqVVDolegz5jxp6gQOW7N5SPxsqLDB5a7YHm5zk0mXSXv5PGsUoeJMd1HZu8QCbFev7is5FerPu8DS9zGKCxt0vKQzCCqWAmEKjfK7MDrfKS9sr9DxYEuffwGVbCJovGE07JhzwF7H4E5cKzJcOE1ajJ4w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Patients’ clinical and laboratory characteristics. (A) FasL-induced apoptosis in patients’ and healthy controls’ phytohemagglutinin- and IL-2–stimulated T-blasts determined by flow cytometry using annexin V/propidium iodide staining showed impaired apoptosis in P1 and P2 before transplantation, normal response in P3 before transplantation, and repaired response in P1 after transplantation compared with healthy control cells. (B) Histopathology of all 3 patients’ lymphoid tumors. P1: EBV+ Hodgkin-like polymorphic B-cell lymphoproliferative disorder; P2: nodal peripheral T-cell lymphoma with follicular helper T-cell phenotype; P3: primary mediastinal large B-cell lymphoma (upper panels [original magnification ×600]: hematoxylin and eosin staining; lower panels [original magnification ×400]: EBV, Epstein-Barr virus encoded RNA in situ hybridization, and CD3 and CD20 immunohistochemical staining). (C) Rapid autologous lymphocyte reconstitution after HSCT in all 3 patients with homozygous TET2 LOF, despite full T-cell–depleting serotherapy. Asterisks indicate the first measurement of T-cell chimerism in each patient: P1 (day +28) 78% recipient; P2 (day +46), 100% recipient; P3 (day +52), 91% recipient.
Patients’ clinical and laboratory characteristics. (A) FasL-induced apoptosis in patients’ and healthy controls’ phytohemagglutinin- and IL-2–stimulated T-blasts determined by flow cytometry using annexin V/propidium iodide staining showed impaired apoptosis in P1 and P2 before transplantation, normal response in P3 before transplantation, and repaired response in P1 after transplantation compared with healthy control cells. (B) Histopathology of all 3 patients’ lymphoid tumors. P1: EBV+ Hodgkin-like polymorphic B-cell lymphoproliferative disorder; P2: nodal peripheral T-cell lymphoma with follicular helper T-cell phenotype; P3: primary mediastinal large B-cell lymphoma (upper panels [original magnification ×600]: hematoxylin and eosin staining; lower panels [original magnification ×400]: EBV, Epstein-Barr virus encoded RNA in situ hybridization, and CD3 and CD20 immunohistochemical staining). (C) Rapid autologous lymphocyte reconstitution after HSCT in all 3 patients with homozygous TET2 LOF, despite full T-cell–depleting serotherapy. Asterisks indicate the first measurement of T-cell chimerism in each patient: P1 (day +28) 78% recipient; P2 (day +46), 100% recipient; P3 (day +52), 91% recipient.
Major clinical features of 3 patients with immunodeficiency and immune dysregulation
| P1 | P2 | P3 | |
|---|---|---|---|
| Immunodeficiency | |||
| Recurrent respiratory tract infections | ++ | + | ++ |
| Bronchiectasis | ++ | + | ++ |
| Herpes viral infection | ++ | + | + |
| Lymphoproliferation | |||
| Lymphadenopathy | + | + | + |
| Hepatosplenomegaly | + | + | + |
| Lymphoma | + | + | + |
| Autoimmunity | |||
| Autoimmune cytopenias | + | + | − |
| Autoantibodies | + | + | − |
| Laboratory values | |||
| Class-switched memory B cells | ↓ | ↓ | ↓ |
| FasL-mediated apoptosis | ↓ | ↓ | Normal |
| Soluble FasL | ↑ | ↑ | Normal |
| DNTs | ↑ | ↑/Normal | ↑ |
| Specific antibodies | Normal | ND | ↓ |
| Developmental delay | |||
| Moderate | + | + | + |
| Outcome of HSCT | |||
| Autologous T-cell reconstitution | + | + | + |
| Split mixed chimerism | Rejected and died | Rejected and died |
| P1 | P2 | P3 | |
|---|---|---|---|
| Immunodeficiency | |||
| Recurrent respiratory tract infections | ++ | + | ++ |
| Bronchiectasis | ++ | + | ++ |
| Herpes viral infection | ++ | + | + |
| Lymphoproliferation | |||
| Lymphadenopathy | + | + | + |
| Hepatosplenomegaly | + | + | + |
| Lymphoma | + | + | + |
| Autoimmunity | |||
| Autoimmune cytopenias | + | + | − |
| Autoantibodies | + | + | − |
| Laboratory values | |||
| Class-switched memory B cells | ↓ | ↓ | ↓ |
| FasL-mediated apoptosis | ↓ | ↓ | Normal |
| Soluble FasL | ↑ | ↑ | Normal |
| DNTs | ↑ | ↑/Normal | ↑ |
| Specific antibodies | Normal | ND | ↓ |
| Developmental delay | |||
| Moderate | + | + | + |
| Outcome of HSCT | |||
| Autologous T-cell reconstitution | + | + | + |
| Split mixed chimerism | Rejected and died | Rejected and died |
+, feature present; ++, major feature; −, feature absent; ↓, lower than normal; ↑, higher than normal; ND, not determined.
Initially, P1 responded well to immunomodulation with high-dose (2 g/kg) IV immunoglobulin, rituximab (anti-CD20 antibody), and corticosteroid. However, at 4 years of age his condition deteriorated markedly with massive hepatosplenomegaly, liver dysfunction, and worsening lymphadenopathy reflecting the development of EBV+ Hodgkin-like polymorphic B-cell lymphoproliferative disorder (Figure 1B; supplemental Figure 1A). Despite hyperhydration, treatment with low-intensity chemotherapy (vincristine and rituximab) led to tumor lysis syndrome with acute renal failure, requiring prolonged intensive care, which was further complicated by Stenotrophomonas pneumonia and sepsis. He received 4 doses of adoptive EBV-specific cytotoxic T cells with therapeutic benefit and underwent splenectomy in preparation for hematopoietic stem cell transplantation (HSCT).
P2 was the younger brother of P1. His health problems began with transient hematuria and nephrotic range proteinuria in the first 4 weeks of life. Subsequently, he was noted to have hypothyroidism and hypogammaglobulinemia that were attributed to renal losses and treated with thyroxine and immunoglobulin supplementation; lymphocyte numbers were normal. He developed cytomegalovirus viremia at ∼8 weeks of age with evidence of respiratory involvement, which was treated with ganciclovir. At around the same age he began to develop hepatosplenomegaly and lymphadenopathy. He was thrombocytopenic, but no autoantibody tests were documented, and Coombs test was negative. However, the clinical diagnosis of ALPS was supported by the demonstration of defective Fas-mediated apoptosis (Figure 1A), variably increased DNTs (1.9%), and increased soluble FasL (0.96 ng/mL) (Table 1). In addition, there was a very low fraction of IgM memory cells (0.33%) and class-switched memory B cells (0.03%). A lymph node biopsy showed a nodal peripheral T-cell lymphoma of follicular helper T-cell (Tfh) phenotype and clonal TCRG gene rearrangement (Figure 1B; supplemental Figure 1B). He was treated with cyclophosphamide and methylprednisolone for his lymphoma; only 1 dose of vincristine was given due to deranged liver function.
P3 was the second child born to related parents from the same community as P1 and P2. Her problems with infection emerged at ∼18 months, with frequent LRTIs requiring multiple courses of antibiotics and, on ≥2 occasions, ventilatory support. There was also a history of loose stools and relatively poor weight gain. Immunologic investigations suggested impaired humoral immunity: she was IgA deficient, with reduced levels of IgM and IgG2 subclass and impaired response to pneumococcal vaccination. Subsequent investigations showed essentially normal lymphocyte subsets, with the exception of missing class-switched memory B cells. Despite immunoglobulin replacement, antibiotic prophylaxis, and physiotherapy, she progressed to bronchiectasis with an overnight oxygen requirement. Furthermore, she showed chronic hepatosplenomegaly and lymphadenopathy, as well as increased DNTs in peripheral blood (9% at age 8 years). A lymph node biopsy showed EBV-associated follicular hyperplasia (supplemental Figure 1C), whereas Fas-dependent apoptosis and T-cell proliferative responses were normal (Figure 1A; Table 1). Although there was no definite evidence of autoimmunity either clinically or serologically, moderate thrombocytopenia was evident over several years. During this time she developed 2 benign skin tumors: a cellular dermatofibroma and a pilomatrixoma (supplemental Figure 1C).
These immunological features occurred against a background of significant global developmental delay; for example, P3 walked at 4 years of age and was not able to attend mainstream school. She had feeding problems and there was a suspicion of recurrent aspiration, managed by fundoplication and creation of a gastrostomy for enteral feeding.
At the age of 12 years, P3 presented with a mediastinal mass and pericardial effusion; investigations revealed a primary mediastinal large B-cell lymphoma (Figure 1B; supplemental Figure 1C). She tolerated R-CHOP chemotherapy (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) and went into remission. At around this time she developed worsening headaches, and idiopathic intracranial hypertension was detected. Imaging revealed skull thickening consistent with extramedullary hematopoiesis.
Early autologous T-cell reconstitution after allogeneic HSCT
After extensive discussions with the family of each patient, and in view of the ongoing risks to health and quality of life, allogeneic HSCT was attempted. Reflecting donor availability and changing clinical practice, each patient received a different conditioning regimen and stem cell source and dose, but outcomes were universally poor (supplemental Table 2). Remarkably, all 3 patients showed autologous T-lymphoid reconstitution within 6 weeks of HSCT, despite a T-cell–depleting conditioning regimen containing full-dose serotherapy (Figure 1C). P2 rejected his haploidentical maternal transplant and died of sepsis, whereas his older brother P1 developed mixed chimerism after a sibling donor transplant, followed by symptomatic relapse of ALPS and disordered hematopoiesis (supplemental Figure 2). P3 tolerated myeloablative conditioning poorly and developed multiorgan failure requiring intensive care (respiratory, renal, circulatory, and gut). Although she survived this phase and went on to receive peripheral blood stem cells from an 11/12 matched unrelated donor, she showed very early reconstitution of autologous T cells with progressive loss of her graft, despite clinical evidence of graft-versus-host disease of skin and liver (grade 3). Treatment was shifted to palliative care at home, where she died.
The clinical and laboratory phenotypes of all 3 patients are outlined in Table 1, supplemental Table 1, and supplemental Figure 3. Details about their HSCTs are provided in supplemental Table 2.
Homozygous LOF mutations of TET2
In view of the striking combination of immunodeficiency, lymphoma, and developmental delay in the setting of consanguinity, an autosomal-recessive inborn error was suspected. WES excluded genes already implicated in ALPS (eg, FAS, FASLG, CASP10, CTLA4, KRAS, MAGT1, NRAS, PIK3CD, STAT3)1 and other known disorders, but it revealed homozygous predicted damaging mutations within the same gene (TET2) in the affected children of both kindreds (supplemental Table 3). Sanger sequencing confirmed that each healthy family member carried ≥1 wt allele of TET2, consistent with autosomal-recessive inheritance (Figure 2A). This gene encodes the epigenetic regulator TET2, a 2-oxoglutarate– and Fe2+-dependent methylcytosine dioxygenase. Somatic mutations in TET2 are strongly linked with CHIP and hematological malignancy (reviewed in Solary et al30 ), making it a strong candidate disease-causing gene in our patients.
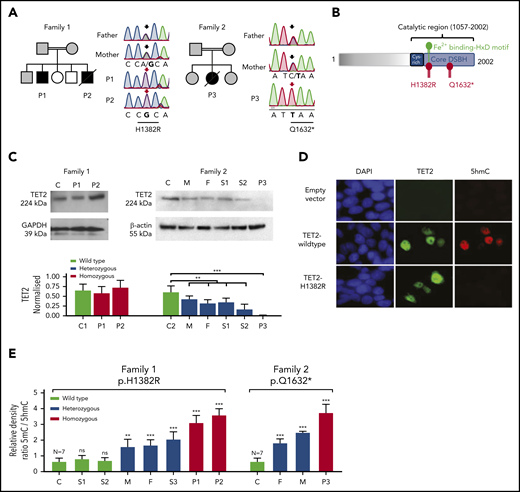
Germline homozygous LOF mutations in TET2 in 3 immunodeficient patients with lymphoma. (A) Pedigree of 2 unrelated consanguineous families, as well as Sanger sequencing results, of patients and unaffected family members with highlighted affected nucleotide (in bold) in the sequence of TET2. Affected patients, all with homozygous variant, are indicated by filled black symbols, heterozygous family members are indicated by gray symbols, and other relatives are indicated by open symbols. Slashes indicate deceased patients. Arrows above Sanger sequencing traces indicate position of mutated nucleotide (black arrow, heterozygous; red arrow, homozygous variant). The effect on amino acid sequence is shown below the underlined mutated codon. (B) Schematic diagram of TET2 protein structure with highlighted mutated residues H1382R and Q1632* within catalytic double stranded β-helix (DSBH) domain, predicting damaging effect of the mutations on enzyme activity. H1382 residue is positioned in the catalytically important Fe2+-binging HxD motif. (C) TET2 protein expression, as detected by immunoblotting, in fibroblasts of an unrelated control (C), P1, and P2 in Family 1 (upper left panel). Absent TET2 expression in P3 and its decreased expression in heterozygous relatives, as detected by immunoblotting, in PBMCs of Family 2: unrelated control (C), mother (M), father (F), sibling 1 (S1), sibling 2 (S2), and P3 (upper right panel). Quantification of TET2 expression normalized to housekeeping proteins GAPDH and β-actin. Data are mean ± SD from 2 independent experiments. **P < .01, ***P < .001 patient vs respective healthy control and pooled heterozygous relatives vs control, unpaired Student t test. (D) Impaired TET2 hydroxymethylating activity detected by 5hmC immunofluorescence staining in HEK293T cells transfected with empty lentiviral vector, Flag-tagged wt TET2, or mutant TET2H1382R. Blue, DAPI stain; green, Flag; red, 5hmC staining. Result is representative of 3 independent experiments. (E) Increased 5mC/5hmC ratio, as determined by DNA methylation assay of total blood DNA, in patients bearing homozygous H1382R and Q1632* mutations (red bars) compared with homozygous wt controls (black bars). Heterozygous relatives (blue bars) showed significantly increased intermediate levels. Data are mean ± SD from 2 independent experiments and 7 healthy controls. **P < .01, ***P < .001 vs healthy controls, unpaired Student t test. ns, not significant.
Germline homozygous LOF mutations in TET2 in 3 immunodeficient patients with lymphoma. (A) Pedigree of 2 unrelated consanguineous families, as well as Sanger sequencing results, of patients and unaffected family members with highlighted affected nucleotide (in bold) in the sequence of TET2. Affected patients, all with homozygous variant, are indicated by filled black symbols, heterozygous family members are indicated by gray symbols, and other relatives are indicated by open symbols. Slashes indicate deceased patients. Arrows above Sanger sequencing traces indicate position of mutated nucleotide (black arrow, heterozygous; red arrow, homozygous variant). The effect on amino acid sequence is shown below the underlined mutated codon. (B) Schematic diagram of TET2 protein structure with highlighted mutated residues H1382R and Q1632* within catalytic double stranded β-helix (DSBH) domain, predicting damaging effect of the mutations on enzyme activity. H1382 residue is positioned in the catalytically important Fe2+-binging HxD motif. (C) TET2 protein expression, as detected by immunoblotting, in fibroblasts of an unrelated control (C), P1, and P2 in Family 1 (upper left panel). Absent TET2 expression in P3 and its decreased expression in heterozygous relatives, as detected by immunoblotting, in PBMCs of Family 2: unrelated control (C), mother (M), father (F), sibling 1 (S1), sibling 2 (S2), and P3 (upper right panel). Quantification of TET2 expression normalized to housekeeping proteins GAPDH and β-actin. Data are mean ± SD from 2 independent experiments. **P < .01, ***P < .001 patient vs respective healthy control and pooled heterozygous relatives vs control, unpaired Student t test. (D) Impaired TET2 hydroxymethylating activity detected by 5hmC immunofluorescence staining in HEK293T cells transfected with empty lentiviral vector, Flag-tagged wt TET2, or mutant TET2H1382R. Blue, DAPI stain; green, Flag; red, 5hmC staining. Result is representative of 3 independent experiments. (E) Increased 5mC/5hmC ratio, as determined by DNA methylation assay of total blood DNA, in patients bearing homozygous H1382R and Q1632* mutations (red bars) compared with homozygous wt controls (black bars). Heterozygous relatives (blue bars) showed significantly increased intermediate levels. Data are mean ± SD from 2 independent experiments and 7 healthy controls. **P < .01, ***P < .001 vs healthy controls, unpaired Student t test. ns, not significant.
P1 and P2 bore the homozygous missense mutation c.4145A>G, p.H1382R in exon 9 of TET2. This was predicted to be highly damaging because it affects the Fe2+-binding motif, which is known to be critical for TET2 enzymatic activity31 (Figure 2B). Furthermore, we identified the same variant in heterozygosity in patients with myeloid malignancy (3/1221 patients with acute myeloid leukemia [AML] and 1/286 patients with chronic myelomonocytic leukemia; supplemental Table 4).32 The expression of TET2H1382R protein was not impaired relative to TET2wt in primary cells (Figure 2C) or in a recombinant system (Figure 2D). We compared the enzymatic activity of TET2wt and TET2H1382R by immunofluorescence microscopy analysis of transfected HEK293T cells stained for 5hmC.22,31 In contrast to TET2wt-overexpressing cells, there was no increase in 5hmC staining in cells expressing mutated TET2H1382R, thus confirming the predicted loss of its 5-hydroxymethylating enzymatic activity (Figure 2D).
The homozygous variant borne by P3 was a nonsense mutation, c.4894C>T, p.Q1632* (Figure 2A). Again, this variant was represented among a cohort of patients with myeloid malignancy (2/1221 patients with AML, 2/286 patients with chronic myelomonocytic leukemia; supplemental Table 4).32 Immunoblotting of PBMCs confirmed loss of TET2 protein expression in the proband, whereas heterozygous relatives showed intermediate levels relative to wt control (Figure 2C).
TET2 LOF results in DNA hypermethylation
We hypothesized that the loss of TET2 enzymatic activity would manifest as an alteration in global DNA methylation/hydroxymethylation status within the hematopoietic compartment. To test this, relative quantification of genomic 5mC and 5hmC was performed in DNA extracted from PBMCs. All patients showed a profound increase in DNA methylation, especially relative to 5hmC. Heterozygous carriers of pathogenic mutations showed a smaller, but measurable, increase in global methylation associated with a reduction in 5hmC (Figure 2E; supplemental Figure 4). We conclude that the TET2 alleles associated with a novel inherited immunodeficiency syndrome produce wide-scale alterations in DNA methylation and hydroxymethylation of circulating leukocytes, with a gene dosage effect.
Altered T-cell homeostasis in TET2 deficiency
All 3 patients showed features of ALPS in the form of variable combinations of hepatosplenomegaly and lymphadenopathy (progressing to lymphoma/lymphoproliferative disorder; n = 3), clinically significant autoimmunity (n = 2), and increased proportion of DNTs (n = 3). Moreover, in P1 and P2, this suggestive clinical picture was accompanied by clearly impaired T-lymphoblast apoptosis in vitro (Figure 1A), together with serologic markers of ALPS, such as elevated levels of soluble CD25, FasL, and IL-10. In contrast, P3 did not show serum markers of ALPS or disordered apoptosis in vitro, despite grossly elevated DNTs (Figure 3A). Surface expression of Fas receptor was normal (data not shown).
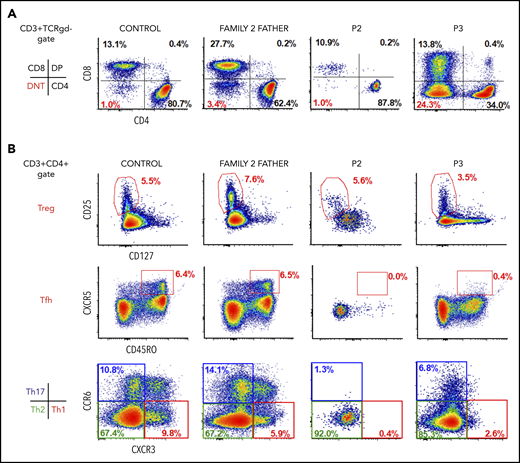
Immunophenotyping of patients and heterozygous relatives. (A) Representative flow cytometry dots plots showing increased levels of DNTs in P3 and normal levels in healthy control, heterozygous relative, and P2. (B) Representative flow cytometry dot plots showing normal levels of Tregs (CD3+CD4+CD25+CD127low) in patients and heterozygous relative, lack of Tfh cells (CD3+CD4+CXCR5+CD45RO+), reduced Th1 cells (CD3+CD4+CCR6−CXCR3+) and Th17 cells (CD3+CD4+CCR6+CXCR3−), and differentiation preference toward Th2 cells (CD3+CD4+CCR6−CXCR3−) in P2 and P3.
Immunophenotyping of patients and heterozygous relatives. (A) Representative flow cytometry dots plots showing increased levels of DNTs in P3 and normal levels in healthy control, heterozygous relative, and P2. (B) Representative flow cytometry dot plots showing normal levels of Tregs (CD3+CD4+CD25+CD127low) in patients and heterozygous relative, lack of Tfh cells (CD3+CD4+CXCR5+CD45RO+), reduced Th1 cells (CD3+CD4+CCR6−CXCR3+) and Th17 cells (CD3+CD4+CCR6+CXCR3−), and differentiation preference toward Th2 cells (CD3+CD4+CCR6−CXCR3−) in P2 and P3.
To examine the effect of TET2 LOF on the differentiation of CD4+ T cells into distinct effector subsets, we performed immunophenotyping of PBMCs from P2 and P3 and their heterozygous relatives using an extensive panel of surface markers. Patients showed decreased T helper 17 (Th17) cell counts (CD3+CD4+CCR6+CXCR3−) and Th1 cell counts (CD3+CD4+CCR6−CXCR3+) and relatively higher Th2 cell counts (CD3+CD4+CCR6−CXCR3−), whereas the proportions of regulatory T cells (Tregs; CD3+CD4+CD25+CD127low) were normal (Figure 3B). Intriguingly, circulating Tfh cells (defined as CD3+CD4+CXCR5+CD45RO+) were nearly absent from TET2-deficient patients. Such a finding is particularly striking given that TET2-mutated T-cell lymphomas, including that in P2, are typically of the Tfh cell type.33 An explanation for this apparent paradox may be that TET2-deficient Tfh cells accumulate in secondary lymphoid tissues, as observed in the spleen of TET2-deficient mice prior to the development of lymphoma.34 Parallel immunophenotyping of PBMCs from heterozygous relatives did not reveal any consistent abnormalities.
TET2 LOF impairs human B-cell terminal differentiation
Although abnormalities in humoral immunity were difficult to disentangle from exogenous immunosuppression in P1, P2 and P3 showed evidence of a primary defect in humoral immunity with a lack of class-switched memory (CD19+CD20+CD27+IgD−) B cells, despite normal overall numbers of B cells and a tendency toward hypergammaglobulinemia (supplemental Table 1). Such a pattern has been observed in other primary immune disorders, such as deficiency in DOCK8 or TPP2.35,36 The lack of surface IgA and IgG, as detected by flow cytometry, confirmed this defect in B-cell differentiation in vivo (Figure 4A).
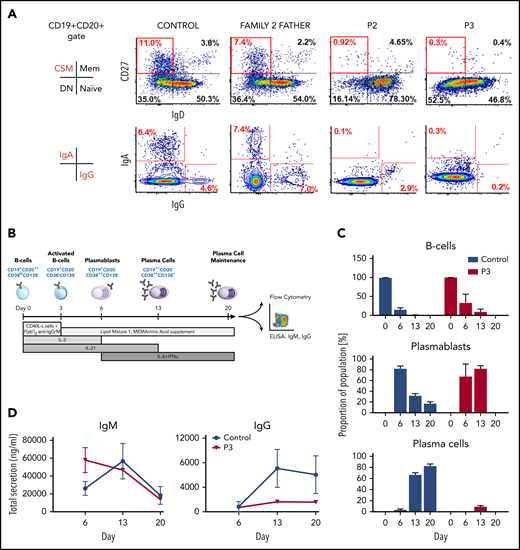
Failure of TET2-deficient B cells to undergo class-switch recombination in vivo, generate mature plasma cells, and produce IgG in vitro. (A) Representative flow cytometry dot plots showing almost absent class-switched memory (CD19+CD20+CD27+IgD−) B cells and block in expression of surface IgA and IgG in patients, as detected in peripheral blood B cells compared with healthy control and family 2 heterozygous relative. (B) Scheme showing in vitro B-cell differentiation strategy after mimicking a T-cell–dependent immune stimulus. (C) Flow cytometric profile of P3 and healthy control in vitro–differentiating primary B cells, indicating a defect in B-cell maturation and impaired cell survival due to loss of TET2 function. (D) Secreted levels of IgM and IgG detected by enzyme-linked immunosorbent assay during B-cell differentiation showed a failure of class-switch recombination in patient cells. Data are mean ± SD from 2 independent experiments.
Failure of TET2-deficient B cells to undergo class-switch recombination in vivo, generate mature plasma cells, and produce IgG in vitro. (A) Representative flow cytometry dot plots showing almost absent class-switched memory (CD19+CD20+CD27+IgD−) B cells and block in expression of surface IgA and IgG in patients, as detected in peripheral blood B cells compared with healthy control and family 2 heterozygous relative. (B) Scheme showing in vitro B-cell differentiation strategy after mimicking a T-cell–dependent immune stimulus. (C) Flow cytometric profile of P3 and healthy control in vitro–differentiating primary B cells, indicating a defect in B-cell maturation and impaired cell survival due to loss of TET2 function. (D) Secreted levels of IgM and IgG detected by enzyme-linked immunosorbent assay during B-cell differentiation showed a failure of class-switch recombination in patient cells. Data are mean ± SD from 2 independent experiments.
To test the hypothesis that TET2 deficiency autonomously impaired the ability of patient B cells to execute an appropriate differentiation program in response to antigenic stimulation, we set up an in vitro culture system that enables the generation of long-lived plasma cells from isolated primary B cells37 (Figure 4B). Phenotypic evaluation of the differentiating populations confirmed that, although TET2Q1632* B cells were capable of initiating plasma cell differentiation, as evidenced by the appearance of short-lived plasmablasts at day 6, these cells failed to progress to phenotypically mature plasma cells, which emerge at day 13 in healthy donors and persist during the time frame of the assay (Figure 4C; supplemental Figure 5). Moreover, assessment of immunoglobulins in the supernatant showed high levels of IgM production by the TET2Q1632* plasmablasts that declined as the cells died and a complete failure to generate IgG (Figure 4D). The plasma cells that survive in this assay have previously been shown to be derived from memory cells that have undergone somatic hypermutation.37 Thus, the lack of IgG is consistent with the scarcity of class-switched memory B cells in the patient and recent data generated in a murine model of TET2 deficiency.38
Acquired somatic mutations in genes within the RAS signaling pathway in patients’ lymphoma tissue
Somatic mutations of TET2 are prevalent within many types of malignancy, including myeloid and lymphoid neoplasms, where they are believed to represent an initiating event39-41 ; therefore, we hypothesized that lymphomagenesis would require a “second hit.” To identify putative cooperating mutations within coding regions, we performed high-depth WES of patients’ lymphoid tumor samples and made a pairwise comparison with germline data, confirming hits by Sanger sequencing.
In the EBV+ Hodgkin-like polymorphic B-cell lymphoproliferative disorder of P1, we detected a single point mutation p.K117N in KRAS, an oncogene that frequently harbors somatic variants in various solid tumor types, as well as hematological malignancies42 (supplemental Table 5). Our observation supports the previously suggested collaboration of TET2 LOF and KRAS gain-of-function mutations, reported in myeloid neoplasia.8,11,43
Another somatic variant, again within the RAS signaling pathway, was found in the peripheral T-cell lymphoma of P2, affecting the gene ERBIN (Erbb2 interacting protein). ERBIN acts within the RAS signaling pathway by disrupting RAS-RAF interaction.44 Our patient’s variant (p.R1194H) had not been reported previously in the context of neoplasia. Predictive models suggest that the mutation may not be deleterious, but we note that it is rare in the population (supplemental Table 5). Unfortunately, we were unable to investigate the acquisition of somatic variants in the primary mediastinal large B-cell lymphoma of P3 because of the lack of material.
TET2 deficiency skews in vitro hematopoietic differentiation toward the myeloid lineage
To study the effect of TET2 hypofunction on human hematopoiesis, we developed an in vitro disease model using patient-derived iPSCs. Primary fibroblasts from P1 and P2 and healthy volunteers were reprogrammed into iPSCs that were fully characterized (supplemental Figure 6) and differentiated into hematopoietic precursors, as described by Olivier et al45 (Figure 5A).

Impaired in vitro hematopoietic differentiation by TET2-deficient iPSCs. (A) Schematic presentation of experimental strategy to assess the hematopoietic differentiation capacity from iPSCs in vitro24 with representative microphotographs (original magnification, ×100) of cell culture at major differentiation stages, starting with embryoid bodies at days 0 to 3, followed by the presence of hematopoietic progenitors (HP) and their budding at days 5 to 12, and culminating in proliferation and maturation at days 13 to 30. (B) Scatter box plots demonstrating the percentage of positive cells detected by flow cytometry at individual time points during differentiation: HP (CD34+/−CD43+), erythro-megakaryocytic progenitors (CD43+CD235a+CD41a+), erythroid progenitors (CD43+CD235a+CD41a−), megakaryocytic progenitors (CD43+CD235a−CD41a+), and myeloid progenitors (CD43+CD235a−CD41a−CD45+). Horizontal lines represent median values from a minimum 6 independent experiments. **P < .01, ***P < .001, nonparametric Kruskal-Wallis test. (C) Quantitative and qualitative results of CFU assay of HP, plated at individual time points of differentiation into semisolid medium and incubated for 2 weeks, showing skewed differentiation toward the myeloid lineage at the expense of erythroid and megakaryocytic (Meg) colonies. Data are mean ± SD from a minimum of 3 independent experiments. (D) Representative microphotographs (original magnification, ×100) of individual CFU classified according to characteristic morphologic features: (i) BFU-E: burst-forming unit-erythoid, (ii) CFU-E: erythroid, (iii) CFU-GM: granulomonocytic, (iv) CFU-Meg: megakaryocytic, (v) above, CFU-E and below, CFU-G: granulocytic, (vi) left, BFU-E and right, CFU-Meg. (E) 5mC/5hmC ratio determined by DNA methylation assay in fibroblasts, iPSCs, and HP from healthy control (C) and patients (P) at day 13 (D13) and day 20 (D20) of hematopoietic differentiation in vitro. Data are mean ± SD from 2 biological repeats. *P < .05 vs healthy control, unpaired Student t test. D9, day 9; D13, day 13; D16, day 16; D20, day 20; D30, day 30; ns, not significant.
Impaired in vitro hematopoietic differentiation by TET2-deficient iPSCs. (A) Schematic presentation of experimental strategy to assess the hematopoietic differentiation capacity from iPSCs in vitro24 with representative microphotographs (original magnification, ×100) of cell culture at major differentiation stages, starting with embryoid bodies at days 0 to 3, followed by the presence of hematopoietic progenitors (HP) and their budding at days 5 to 12, and culminating in proliferation and maturation at days 13 to 30. (B) Scatter box plots demonstrating the percentage of positive cells detected by flow cytometry at individual time points during differentiation: HP (CD34+/−CD43+), erythro-megakaryocytic progenitors (CD43+CD235a+CD41a+), erythroid progenitors (CD43+CD235a+CD41a−), megakaryocytic progenitors (CD43+CD235a−CD41a+), and myeloid progenitors (CD43+CD235a−CD41a−CD45+). Horizontal lines represent median values from a minimum 6 independent experiments. **P < .01, ***P < .001, nonparametric Kruskal-Wallis test. (C) Quantitative and qualitative results of CFU assay of HP, plated at individual time points of differentiation into semisolid medium and incubated for 2 weeks, showing skewed differentiation toward the myeloid lineage at the expense of erythroid and megakaryocytic (Meg) colonies. Data are mean ± SD from a minimum of 3 independent experiments. (D) Representative microphotographs (original magnification, ×100) of individual CFU classified according to characteristic morphologic features: (i) BFU-E: burst-forming unit-erythoid, (ii) CFU-E: erythroid, (iii) CFU-GM: granulomonocytic, (iv) CFU-Meg: megakaryocytic, (v) above, CFU-E and below, CFU-G: granulocytic, (vi) left, BFU-E and right, CFU-Meg. (E) 5mC/5hmC ratio determined by DNA methylation assay in fibroblasts, iPSCs, and HP from healthy control (C) and patients (P) at day 13 (D13) and day 20 (D20) of hematopoietic differentiation in vitro. Data are mean ± SD from 2 biological repeats. *P < .05 vs healthy control, unpaired Student t test. D9, day 9; D13, day 13; D16, day 16; D20, day 20; D30, day 30; ns, not significant.
TET2H1382R cultures showed a higher proportion of erythro-megakaryocytic progenitors and a persistently smaller fraction of myeloid progenitors, as defined by surface marker expression (Figure 5B; supplemental Figure 7A-F). Yet, paradoxically, CFU assay revealed a skewed and boosted clonogenic potential of TET2H1382R hematopoietic progenitors toward the myeloid lineage, whereas erythroid and megakaryocytic colony formation was severely impaired (Figure 5C-D). Our observation is in agreement with a previous study showing hyperproliferation and impaired differentiation of TET2-deficient erythroid cells in mice.46 This could be correlated with in vivo findings of marked monocytosis and variable neutrophilia in patients P1 and P3, whereas all 3 patients showed chronic thrombocytopenia (supplemental Figure 1). Moreover, the surviving patient (P1) has developed a transfusion-dependent anemia over time, although we cannot rule out a late effect of chemotherapy on his marrow reserve.
Measurement of DNA methylation status in patients’ fibroblasts, iPSCs, and hematopoietic progenitors revealed DNA hypermethylation in nondifferentiated iPSCs and early-stage hematopoietic progenitors at days 13 and day 20 (Figure 5E; supplemental Figure 4C). This corresponds to the time at which TET2 expression is upregulated during hematopoietic differentiation (supplemental Figure 7G) and echoes the DNA hypermethylation detected in patients’ whole blood (Figure 2E). We saw no evidence of compensation of TET2 deficiency by upregulation of other TET family members (supplemental Figure 7G), in keeping with observations in TET2-knockout (KO) mice.28,47
Discussion
This is the first report of human germline homozygous TET2 LOF, which we identified in association with combined immunodeficiency, autoimmunity, and childhood lymphoma in 2 unrelated kindreds. In keeping with a severe autosomal-recessive trait, inborn homozygous null mutations of this gene are absent from databases of human genomic variation.48,49 There are 2 recent reports of inherited heterozygous deficiency of TET2 in humans. In a family with lymphoma, Kaasinen et al found carriers (some, but not all, of whom were affected) of a heterozygous germline mutation that had already been reported as a somatic mutation in an AML patient, and 1 additional unrelated case with a de novo variant showing mild developmental delay.17 Subsequently, Duployez et al described 3 siblings with myeloid malignancy in the sixth and seventh decades of life and a shared heterozygous germline frameshift mutation in TET2; their mother had died of T-cell lymphoma, but no DNA was available from her.50 To our knowledge, the extensive literature on human TET2 deficiency focuses exclusively on the more frequent somatic variations, both heterozygous and homozygous, mainly in the context of CHIP, myeloid, and lymphoid malignancies.8,9,51 Our findings confirm the strong link between TET2 LOF and lymphomagenesis, with early-onset lymphoid tumors of diverse types in all 3 affected children. At present, we must consider healthy heterozygous family members as being at increased risk for hematologic malignancy, but this is difficult to quantify.
Our patients’ phenotype is also consistent with observations of TET2-KO mice that develop normally but, nevertheless, demonstrate myeloproliferation, splenomegaly, and lymphomagenesis. Indeed, engineered TET2−/− mice have earlier onsets of myeloproliferative neoplasia than do TET2+/− mice that exhibit similar, but less severe, symptoms.24,27 The TET2 defect increases blood DNA methylation levels, especially at active enhancers and cell type–specific regulatory regions with binding sequences of master transcription factors involved in hematopoiesis, endorsing the importance of TET2 in the regulation of hematopoietic differentiation.17-19 Furthermore, 5hmC level in DNA was reduced dramatically in homozygous TET2-mutant mice compared with heterozygotes,27 as likewise noticed in peripheral blood in our cohort: patient-derived iPSCs and hematopoietic progenitors generated in vitro. Dominguez et al. related murine TET2 LOF to specific alterations in DNA hydroxymethylation and corresponding gene expression within germinal center B cells.38 Interestingly, this effect was seen only when TET2 was absent at earlier stages of B-cell differentiation, implying epigenetic imprinting.
Our patients’ immunodeficiency and immune dysregulation emphasize a broader role for TET2 in homeostasis and function of the human adaptive immune system. Although an immunodeficiency phenotype has not been reported for Tet2-KO mice, recent studies imply a crucial role for TET2 in maintaining T-cell homeostasis and B-cell development.38,52,53 In agreement with the mouse KO model, in which TET2 deficiency and subsequent 5hmC reduction affected differentiation of T-cell subsets,54 we detected relative loss of Th subsets (Tfh, Th1, and Th17), with a skew toward the Th2 fate and preservation of Treg numbers. Abnormal Treg proliferation and Foxp3 destabilization were observed only after double Tet2/Tet3-KO in mice.52,55 Moreover, TET2-deficient B cells in both species display deficiencies in the generation of memory populations, as well as mature plasma cells, thereby compromising long-term effective humoral immunity. That this should cause an immunodeficiency phenotype in human beings in the natural environment is, perhaps, not surprising.
It is noteworthy that clinically relevant autoimmunity and impaired T-cell apoptosis in our TET2H1382R patients cosegregated and that these abnormalities were absent from P3 and the KO mouse model. One attractive hypothesis is that the hypomorphic nature of the H1382R mutation dissociates the enzymatic and nonenzymatic epigenetic activities of TET2,56,57 potentially modulating disease phenotype. However, it is by no means uncommon for individual IEIs to produce a broad disease spectrum, ranging from lymphoproliferation to immunodeficiency.1 We can conclude that impaired T-cell apoptosis shows variable expressivity in TET2 deficiency, but at present we cannot confidently ascribe this to a genotype-phenotype effect. Indeed, all of the patients reconstituted autologous T cells strikingly early after conditioned HSCT, suggesting a cell-autonomous proproliferative phenotype. This is in keeping with the observed outgrowth of a clone of chimeric antigen receptor T cells with biallelic TET2 inactivation in a case of successful cancer immunotherapy58 and expanded CD8 memory response after lymphocytic choriomeningitis virus challenge in a mouse model.59
We did not find any impairment in the reprogramming efficiency or pluripotent potential of TET2-deficient iPSCs, just as mouse embryonic stem cells deleted for TET proteins retain pluripotency.60,61 Increased hematopoietic repopulating capacity with skewing of cell differentiation toward monocytic/granulocytic lineages was described in Tet2-KO mice that died by 1 year of age because of the development of myeloid malignancies.27 Our results from in vitro hematopoietic differentiation of TET2-deficient iPSCs confirm a similar effect in humans, showing boosted clonogenic potential of myeloid progenitors at the expense of impaired erythroid and megakaryocytic progenitors. There were echoes of this in vivo in the 2 patients who survived infancy, both of whom showed coexistent monocytosis and frequent neutrophilia, along with thrombocytopenia.
Had sufficient material been available, it would have been ideal to explore our observations further at transcriptomic and epigenomic levels. Such analysis awaits the identification of future cases, which might now be achieved by targeted screening among children with lymphoid malignancy, especially on a background of consanguinity and immunodeficiency. Nonetheless, the present findings expand our understanding of the critical role of TET2 within the human hematopoietic system and define a new IEI.
Data sharing requests should be sent to Sophie Hambleton (sophie.hambleton@newcastle.ac.uk).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and their families, as well as clinical colleagues at the Great North Children’s Hospital, Leeds General Infirmary, and Pinderfields Hospital, Wakefield. They acknowledge support from the flow cytometry core, and bioimaging and genomics facilities at Newcastle University and the University of Leeds. They thank Skirmantas Kraucionis for reagents and Richard Cornall and Consuelo Anzilotti for helpful discussions.
This work was supported by the Sir Jules Thorn Charitable Trust (12/JTA), the Newcastle NIHR Biomedical Research Centre, the Newcastle upon Tyne Hospitals Charity, and the Wellcome Trust (207556/Z/17/Z) (all S.H.); Leeds Cares charity and Leeds NIHR Biomedical Research Centre (S.S.); and Leukaemia and Lymphoma Research (09005) (M.L.).
Authorship
Contribution: J.S.S. and D.L., and N.V.M. designed and performed experiments, analyzed data, and wrote the manuscript; S.M.B.M., A.R.-E., S.E., F.R.-L., C.C., and S.P. performed experiments and analyzed data; K.R.E. designed experiments and wrote the manuscript; A.M. and H.G. performed bioinformatics analyses; M.A. assisted with histopathology; G.D., J.S., J.A.P., and R.A. designed and performed experiments and analyzed data; A.J.C., K.W., P.C., S.O., E.G.S., and C.R.C. cared for patients and provided clinical data; M.L. designed experiments; C.M.B. analyzed histopathology, designed and performed experiments, and analyzed data; and S.S. and S.H. cared for patients, provided clinical data, designed and analyzed experiments, and wrote the manuscript. All authors read and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sophie Hambleton, Newcastle University Translational and Clinical Research Institute, Leech Building #M3.136D, Framlington Place, Newcastle upon Tyne NE2 4HH, United Kingdom; e-mail: sophie.hambleton@newcastle.ac.uk; and Sinisa Savic, Leeds Institute of Rheumatic and Musculoskeletal Medicine, Wellcome Trust Brenner Building, St James's University Hospital, Leeds LS9 7TF, United Kingdom, e-mail: s.savic@leeds.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal