

Key Points
Decitabine systemic exposures, pharmacodynamics, and safety with oral cedazuridine/decitabine were similar to IV decitabine.
Efficacy and safety from oral cedazuridine/decitabine were consistent with those from IV decitabine in MDS and CMML patients.

Abstract
This phase 2 study was designed to compare systemic decitabine exposure, demethylation activity, and safety in the first 2 cycles with cedazuridine 100 mg/decitabine 35 mg vs standard decitabine 20 mg/m2 IV. Adults with International Prognostic Scoring System intermediate-1/2- or high-risk myelodysplastic syndromes (MDS) or chronic myelomonocytic leukemia (CMML) were randomized 1:1 to receive oral cedazuridine/decitabine or IV decitabine in cycle 1, followed by crossover to the other treatment in cycle 2. All patients received oral cedazuridine/decitabine in subsequent cycles. Cedazuridine and decitabine were given initially as separate capsules in a dose-confirmation stage and then as a single fixed-dose combination (FDC) tablet. Primary end points: mean decitabine systemic exposure (geometric least-squares mean [LSM]) of oral/IV 5-day area under curve from time 0 to last measurable concentration (AUClast), percentage long interspersed nuclear element 1 (LINE-1) DNA demethylation for oral cedazuridine/decitabine vs IV decitabine, and clinical response. Eighty patients were randomized and treated. Oral/IV ratios of geometric LSM 5-day AUClast (80% confidence interval) were 93.5% (82.1-106.5) and 97.6% (80.5-118.3) for the dose-confirmation and FDC stages, respectively. Differences in mean %LINE-1 demethylation between oral and IV were ≤1%. Clinical responses were observed in 48 patients (60%), including 17 (21%) with complete response. The most common grade ≥3 adverse events regardless of causality were neutropenia (46%), thrombocytopenia (38%), and febrile neutropenia (29%). Oral cedazuridine/decitabine (100/35 mg) produced similar systemic decitabine exposure, DNA demethylation, and safety vs decitabine 20 mg/m2 IV in the first 2 cycles, with similar efficacy. This study is registered at www.clinicaltrials.gov as #NCT02103478.
Introduction
The cytidine-nucleoside analogs decitabine and azacitidine are DNA methyltransferase (DNMT) inhibitors (hypomethylating agents [HMAs]) that incorporate into DNA during the S-phase of the cell cycle, thereby reducing methylation of cytosine-phosphate-guanine dinucleotide residues in genomic DNA and, in turn, modifying epigenetic patters and gene expression.1,2 The inhibition of DNA methylation and subsequent gene reexpression is believed to contribute to the clinical activity of these agents,3,4 which are used in the treatment of patients with myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia (CMML),5-8 and patients with acute myeloid leukemia (AML) who are not candidates for high-intensity therapy.9,10 Both agents are administered parenterally for 5 to 7 days/treatment cycle and multiple cycles are generally needed for maximal clinical response.6,11-13 An orally bioavailable DNMT inhibitor would provide a therapeutic advance over current therapy and the potential to improve quality of life by allowing home treatment and reducing the burden associated with monthly, multiple-day IV or subcutaneous treatment in a clinic/hospital setting. This is especially important for long-term responders who are treated over prolonged periods and may benefit the most. The oral bioavailability of decitabine and azacitidine is limited because of rapid inactivation by cytidine deaminase (CDA) in the gastrointestinal (GI) tract and liver.14-16 High oral doses of azacitidine (up to 600 mg) are required to achieve modest systemic exposure (maximum 20% bioavailability), but are associated with significant GI toxicity (grade 3/4 diarrhea in 12% of patients) and high variability in systemic exposures.15
Inhibition of CDA represents a viable approach to improving the oral bioavailability of DNMT inhibitors. The competitive CDA inhibitor tetrahydrouridine (THU) increases the oral bioavailability of decitabine, but THU is unstable in acidic environments, making it pharmaceutically difficult to develop.17,18 Cedazuridine (E7727; Astex Pharmaceuticals, Inc., Pleasanton, CA), a novel CDA inhibitor designed to overcome the instability of THU, safely and effectively increased decitabine exposure following oral administration in preclinical studies.17,19 The first-in-human dose-escalation trial of concurrently administered oral cedazuridine plus decitabine at various doses produced decitabine exposure and DNA demethylation comparable to IV decitabine at doses of cedazuridine 100 mg and decitabine 30 to 40 mg.20 Herein, we present results of a phase 2 study with the selected oral doses of cedazuridine 100 mg and decitabine 35 mg compared with IV decitabine 20 mg/m2.
Patients and methods
Study design and patients
This phase 2, multicenter, open-label, randomized, crossover study was designed to compare the pharmacokinetics (PK), pharmacodynamics (PD) of DNA demethylation, and safety of oral cedazuridine/decitabine (ASTX727; Astex Pharmaceuticals, Inc.) with IV decitabine in the first 2 randomized treatment cycles, and then to assess the long-term efficacy and safety of oral cedazuridine/decitabine after long-term treatment with the oral drug as a single arm. Eligible patients were initially randomized 1:1 to receive 1 of 2 treatment sequences during the first 2 28-day cycles: oral cedazuridine/decitabine daily for 5 days in cycle 1, followed by IV decitabine daily for 5 days in cycle 2 (sequence A); or IV decitabine in cycle 1, followed by the oral drug in cycle 2 (sequence B).
Major eligibility criteria were age ≥18 years, intermediate-1/2- or high-risk MDS by the International Prognostic Scoring System, or CMML, Eastern Cooperative Oncology Group performance status 0 to 2, adequate hepatic (≤2× upper limit of normal [ULN] for bilirubin, and ≤2.5× ULN for aspartate and alanine aminotransferase) and renal (≤1.5× ULN for serum creatinine or >50 mL/min per 1.73 m2) function, and no evidence of active second malignancy. One prior cycle of either decitabine or azacitidine was allowed, but no other cytotoxic chemotherapy was permitted within 2 weeks of starting study treatment. Patients with prior allogeneic hematopoietic cell transplants were eligible as long as they were free of graft-versus-host disease and off immunosuppressive therapy at the time of enrollment.
The primary end points of the study were oral/IV decitabine exposure over 5 days, DNA demethylation of oral cedazuridine/decitabine vs IV decitabine from the first 2 cycles, and overall response rate using International Working Group 2006 criteria.21 Secondary end points included other efficacy outcomes (duration of response, transfusion independence, time to AML, and survival), other PK measurements, and assessment of the safety of oral cedazuridine/decitabine vs IV decitabine in the first 2 cycles, and of the oral drug from cycle 3 onwards.
The protocol was approved by the institutional review board at each study center before any study-related procedures were conducted, and the study was conducted in accordance with Good Clinical Practice guidelines, local regulatory requirements, and ethical principles enunciated in the Declaration of Helsinki. All patients provided written informed consent.
Treatment
Patients were initially randomized to receive oral cedazuridine 100 mg and decitabine 35 mg in fasting conditions daily for 5 days in cycles 1 (sequence A) or 2 (sequence B); or IV decitabine at the standard dose of 20 mg/m2 per day for 5 days by 1-hour infusion in cycles 1 (sequence B) or 2 (sequence A). All patients received oral treatment from cycle 3 onwards. Initially, patients received the 2 oral drugs concomitantly as separate capsules in a first stage for confirmation of the selected doses (dose-confirmation stage). After preliminary PK analyses in this cohort showed comparable decitabine exposure of oral and IV decitabine, a second cohort was randomized using the fixed-dose combination (FDC) tablet containing the 2 drugs at the same doses (FDC stage). Cycles were repeated every 28 days. Dose delay at the discretion of the investigator was permitted to allow for count recovery in case of drug-related myelosuppression. Dose reduction was not allowed in the first 2 cycles, but was permitted from cycle 3 onwards by reducing the number of days of oral treatment. All patients received treatment until disease progression, unacceptable toxicity, or withdrawal by patient or investigator for other reasons.
PK measures
Peripheral blood samples (3 mL) for PK analysis were collected and stored in the specified protocol conditions. For oral study treatment, serial blood samples from predose until 24 hours postdose were collected on days 1, 2, and 5 from the dose-confirmation cohort, and days 1 and 5 from the FDC cohort once it was determined that exposures in days 2 and 5 were similar. For IV decitabine, blood samples were collected serially on day 1 predose until 8 hours postdose. Plasma samples were analyzed for concentrations of decitabine, cedazuridine, and cedazuridine-epimer using a validated liquid chromatography–tandem mass spectrometry method at Frontage Labs (Exton, PA).
Pharmacodynamic measures
DNA methylation was assessed using the long interspersed nuclear element 1 (LINE-1) methylation bisulfite sequencing assay, as previously reported.22 Blood samples for assessing DNA methylation were collected at screening, predose on day 1 of cycles 1, 2, and 3, and days 8, 15, and 22 of cycles 1 and 2. Changes in DNA methylation after treatment were expressed as relative percent change from baseline, as previously described.20 Baseline was defined as the last value obtained predose on day 1 of cycles 1 and 2.
Efficacy and safety measures
Peripheral blood counts were obtained weekly in the first 2 cycles and then at least on day 1 of each subsequent cycle. Bone marrow aspirate or biopsy was performed every 2 cycles for response assessment. Safety was assessed by patient-reported and investigator-observed adverse events (AEs), clinical laboratory testing, physical examination, and electrocardiogram. Clinical chemistry and 12-lead electrocardiogram were obtained on day 1 of each cycle. Adverse events were reported using the Common Terminology Criteria for Adverse Events v4.0.
Sample size and statistical analysis
Sample size was estimated separately for the dose-confirmation and FDC stages. For the dose-confirmation cohort, in an equivalence test of the mean decitabine 5-day area under the plasma concentration-time curve (AUC) of oral cedazuridine/decitabine vs IV decitabine using 2 1-sided tests on data from a 2 × 2 (2-cycle, 2-sequence) crossover design, a sample size of 42 evaluable patients achieved 86% power at a 10% significance level when the true ratio of the means was 1.0, the coefficient of variation on the original scale was 0.5, and the equivalence limits of the mean ratio were 0.75 and 1.33. For the FDC cohort, a total of 18 to 24 evaluable patients provided 75% to 88% power at a 10% significance level when the true ratio of the means was 1.0, the coefficient of variation on the original scale was 0.55, and the equivalence limits for the ratio of means were 0.65 and 1.539. To compensate for nonevaluable patients for the PK analyses, approximately 50 and 30 patients were allowed to be treated in the dose-confirmation and FDC cohorts, respectively.
All statistical tests were conducted using SAS 9.4 (SAS Institute, Cary, NC). The PK analysis of the concentration-time data was performed by noncompartmental methods with Phoenix WinNonlin by Certara Strategic Consulting (Montreal, QC, Canada).
The primary oral/IV AUC from time 0 to last measurable concentration (AUClast) analysis was conducted in patients who successfully received and provided sufficient PK samples from the first 2 randomized cycles of oral and IV decitabine (primary paired population). Secondary exposure analyses including AUC from time 0 to 24 hours postdose and to infinity were also performed for all patients who received ≥1 cycle of treatment (unpaired population). Analysis of variance (ANOVA) was performed on natural log-transformed decitabine 5-day AUClast. Equivalence between treatments was achieved if the ratio of the geometric least-squares mean (LSM) and its 80% confidence interval (CI) were fully contained within the prespecified CI limits of 75 to 133 in the dose-confirmation cohort and 65 to 153.9 in the FDC cohort.
For AUC, comparisons between oral and IV treatments were conducted independently for each of the dose-confirmation and FDC cohorts because they used a different drug product formulation. For all other PD, efficacy, and safety analyses, comparisons were made for the overall patient population because all patients in the study received the same treatment doses (cedazuridine 100 mg/decitabine 35 mg) or decitabine 20 mg/m2 IV.
The PD analysis set for DNA demethylation included all patients who received ≥1 cycle of study treatment, and had baseline and day 8 or 15 LINE-1 demethylation data. Comparisons were performed separately for each cycle to avoid the carryover effect of DNA methylation from cycles 1 to 2. Maximum %LINE-1 demethylation, with 95% CIs for the difference in mean maximum %LINE-1 demethylation between oral and IV treatments, was generated using ANOVA separately for cycles 1 and 2.
Safety and efficacy were assessed and summarized descriptively in all patients who received any study treatment. Time-to-event data were assessed using the Kaplan-Meier method. The study protocol encompassed both phases 1 and 2.
Results
At the data cutoff for this report, the median follow-up was 24.3 months (range, 12.0-29.2 months).
Patients
In all, 138 patients were screened for participation and 86 were randomized, including 52 and 34 into the dose-confirmation and FDC cohorts, respectively (Figure 1). Of these patients, 6 did not receive any study treatment (2 and 4 in the dose-confirmation and FDC cohorts, respectively) and were excluded from all analyses. At data cutoff, 67 patients had discontinued treatment and 13 remained on treatment. Patient characteristics are shown in Table 1. Baseline characteristics were generally balanced across the first 2 randomized treatment sequences in each of the 2 cohorts.
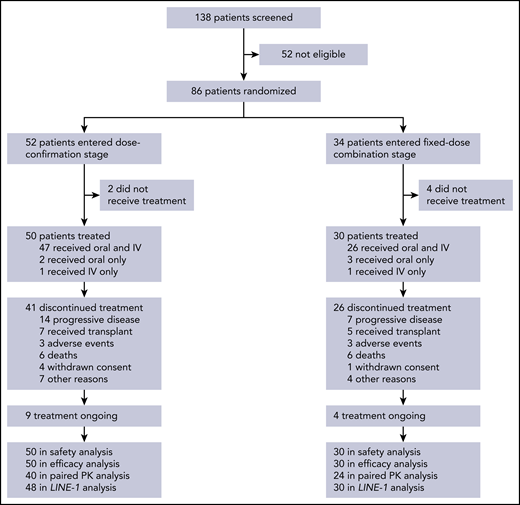
Patient disposition. Six patients did not receive study treatment, including 2 who became ineligible because of elevated liver enzymes, 1 with progressive disease, 1 who died, 1 misdiagnosed, and 1 who withdrew consent after randomization and before start of treatment.
Patient disposition. Six patients did not receive study treatment, including 2 who became ineligible because of elevated liver enzymes, 1 with progressive disease, 1 who died, 1 misdiagnosed, and 1 who withdrew consent after randomization and before start of treatment.
Patient characteristics
| Characteristic | DC cohort (separate capsules) | FDC cohort (FDC tablet) | Phase 2 overall (N = 80) | ||||
|---|---|---|---|---|---|---|---|
| Sequence A (n = 25) | Sequence B (n = 25) | Total (n = 50) | Sequence A (n = 16) | Sequence B (n = 14) | Total (n = 30) | ||
| Mean age, y (SD) | 69.3 (11.2) | 70.2 (10.5) | 69.7 (10.7) | 69.9 (12.1) | 69.4 (9.0) | 69.6 (10.6) | 69.7 (10.6) |
| Median (range) | 69 (32-87) | 72 (41-86) | 71.5 (32-87) | 71 (40-90) | 70 (53-82) | 70.5 (40-90) | 71 (32-90) |
| Sex | |||||||
| Male | 20 (80) | 21 (84) | 41 (82) | 12 (75) | 8 (57) | 20 (67) | 61 (76) |
| Female | 5 (20) | 4 (16) | 9 (18) | 4 (25) | 6 (43) | 10 (33) | 19 (24) |
| Ethnic origin | |||||||
| White | 24 (96) | 22 (88) | 46 (92) | 14 (88) | 14 (100) | 28 (93) | 74 (93) |
| Black or African American | 1 (4) | 1 (4) | 2 (4) | 0 (0) | 0 (0) | 0 (0) | 2 (2.5) |
| Other | 0 (0) | 2 (8) | 2 (4) | 2 (12) | 0 (0) | 2 (7) | 4 (5) |
| Median weight, kg (range) | 82 (40-122) | 87 (55-118) | 85 (40-122) | 76 (49-100) | 83 (42-98) | 80 (42-100) | 83 (40-122) |
| Mean BSA, m2 (SD) | 1.9 (0.3) | 2.0 (0.2) | 2.0 (0.3) | 1.9 (0.2) | 1.9 (0.3) | 1.9 (0.2) | 2.0 (0.3) |
| ECOG performance status | |||||||
| 0 | 13 (52) | 9 (36) | 22 (44) | 7 (44) | 6 (43) | 13 (43) | 35 (44) |
| 1 | 9 (36) | 15 (60) | 24 (48) | 9 (56) | 5 (36) | 14 (47) | 38 (48) |
| 2 | 3 (12) | 1 (4) | 4 (8) | 0 (0) | 3 (21) | 3 (10) | 7 (9) |
| Disease and IPSS category | |||||||
| MDS intermediate 1 | 10 (40) | 10 (40) | 20 (40) | 9 (56) | 6 (43) | 15 (50) | 35 (44) |
| MDS intermediate 2 | 6 (24) | 7 (28) | 13 (26) | 3 (19) | 3 (21) | 6 (20) | 19 (24) |
| MDS high risk | 4 (16) | 4 (16) | 8 (16) | 1 (6) | 0 (0) | 1 (3) | 9 (11) |
| CMML | 5 (20) | 4 (16) | 9 (18) | 3 (19) | 5 (36) | 8 (27) | 17 (21) |
| Prior HMA | 2 (8) | 1 (4) | 3 (6) | 1 (6) | 3 (21) | 4 (13) | 7 (9) |
| RBC transfusion dependence | 9 (36) | 13 (52) | 22 (44) | 11 (69) | 5 (36) | 16 (53) | 38 (47) |
| >5% bone marrow blasts | 14 (61) | 14 (56) | 28 (58) | 8 (53) | 5 (36) | 13 (45) | 41 (53) |
| Characteristic | DC cohort (separate capsules) | FDC cohort (FDC tablet) | Phase 2 overall (N = 80) | ||||
|---|---|---|---|---|---|---|---|
| Sequence A (n = 25) | Sequence B (n = 25) | Total (n = 50) | Sequence A (n = 16) | Sequence B (n = 14) | Total (n = 30) | ||
| Mean age, y (SD) | 69.3 (11.2) | 70.2 (10.5) | 69.7 (10.7) | 69.9 (12.1) | 69.4 (9.0) | 69.6 (10.6) | 69.7 (10.6) |
| Median (range) | 69 (32-87) | 72 (41-86) | 71.5 (32-87) | 71 (40-90) | 70 (53-82) | 70.5 (40-90) | 71 (32-90) |
| Sex | |||||||
| Male | 20 (80) | 21 (84) | 41 (82) | 12 (75) | 8 (57) | 20 (67) | 61 (76) |
| Female | 5 (20) | 4 (16) | 9 (18) | 4 (25) | 6 (43) | 10 (33) | 19 (24) |
| Ethnic origin | |||||||
| White | 24 (96) | 22 (88) | 46 (92) | 14 (88) | 14 (100) | 28 (93) | 74 (93) |
| Black or African American | 1 (4) | 1 (4) | 2 (4) | 0 (0) | 0 (0) | 0 (0) | 2 (2.5) |
| Other | 0 (0) | 2 (8) | 2 (4) | 2 (12) | 0 (0) | 2 (7) | 4 (5) |
| Median weight, kg (range) | 82 (40-122) | 87 (55-118) | 85 (40-122) | 76 (49-100) | 83 (42-98) | 80 (42-100) | 83 (40-122) |
| Mean BSA, m2 (SD) | 1.9 (0.3) | 2.0 (0.2) | 2.0 (0.3) | 1.9 (0.2) | 1.9 (0.3) | 1.9 (0.2) | 2.0 (0.3) |
| ECOG performance status | |||||||
| 0 | 13 (52) | 9 (36) | 22 (44) | 7 (44) | 6 (43) | 13 (43) | 35 (44) |
| 1 | 9 (36) | 15 (60) | 24 (48) | 9 (56) | 5 (36) | 14 (47) | 38 (48) |
| 2 | 3 (12) | 1 (4) | 4 (8) | 0 (0) | 3 (21) | 3 (10) | 7 (9) |
| Disease and IPSS category | |||||||
| MDS intermediate 1 | 10 (40) | 10 (40) | 20 (40) | 9 (56) | 6 (43) | 15 (50) | 35 (44) |
| MDS intermediate 2 | 6 (24) | 7 (28) | 13 (26) | 3 (19) | 3 (21) | 6 (20) | 19 (24) |
| MDS high risk | 4 (16) | 4 (16) | 8 (16) | 1 (6) | 0 (0) | 1 (3) | 9 (11) |
| CMML | 5 (20) | 4 (16) | 9 (18) | 3 (19) | 5 (36) | 8 (27) | 17 (21) |
| Prior HMA | 2 (8) | 1 (4) | 3 (6) | 1 (6) | 3 (21) | 4 (13) | 7 (9) |
| RBC transfusion dependence | 9 (36) | 13 (52) | 22 (44) | 11 (69) | 5 (36) | 16 (53) | 38 (47) |
| >5% bone marrow blasts | 14 (61) | 14 (56) | 28 (58) | 8 (53) | 5 (36) | 13 (45) | 41 (53) |
Data are n (%) unless otherwise specified.
BSA, body surface area; DC, dose confirmation; FDC, fixed dose combination; ECOG, Eastern Cooperative Oncology Group; IPSS, International Prognostic Scoring System; RBC, red blood cell; SD, standard deviation.
Treatment
Patients received a median of 7 treatment cycles (range, 1-29). Thirty-two patients (40%) had ≥1 dose reduction and 41 patients (51%) had ≥1 cycle delayed.
Pharmacokinetics
Table 2 shows the PK AUC results: 40/50 and 24/30 patients treated in the dose-confirmation and FDC cohorts, respectively, had paired oral and IV PK data from the first 2 randomized cycles to calculate AUClast, and were included in the primary end point analysis of 5-day decitabine AUClast. The 5-day decitabine AUClast oral/IV geometric LSM ratios were 93.5 (80% CI, 82.1-106.5) and 97.6 (80% CI, 80.5-118.3) in the dose-confirmation and FDC cohorts, respectively. In both cohorts oral/IV decitabine AUC exposures were within the prespecified 80% CI limits. This conclusion was supported by secondary analyses of the different 5-day AUC parameters in the unpaired PK populations (including additional patients who had sufficient PK data from 1 cycle).
Decitabine AUC for oral cedazuridine/decitabine vs IV decitabine
| Parameter | IV geometric LSM | Oral geometric LSM | LSM ratio (oral/IV) | 80% CI | Intrapatient CV% |
|---|---|---|---|---|---|
| Primary paired population | |||||
| 5-d AUClast, ng × hr per mL (primary end point) | |||||
| DC cohort (n = 40) | 802.81 | 750.82 | 93.52 | 82.10-106.5 | 47.0 |
| FDC cohort (n = 24) | 745.26 | 727.29 | 97.59 | 80.48-118.3 | 53.8 |
| Secondary unpaired population | |||||
| 5-d AUClast, ng × hr per mL | |||||
| DC cohort | 795.41 (n = 42) | 735.62 (n = 48) | 92.48 | 81.37-105.1 | 48.4 |
| FDC cohort | 742.26 (n = 26) | 760.43 (n = 28) | 102.45 | 85.35-123.0 | 52.7 |
| 5-d AUC24, h/ng per mL | |||||
| DC cohort | 794.73 (n = 40) | 753.68 (n = 45) | 94.83 | 83.97-107.1 | 43.5 |
| FDC cohort | 696.90 (n = 20) | 846.82 (n = 26) | 121.51 | 97.15-152.0 | 59.1 |
| 5-d AUC∞, ng × hr per mL | |||||
| DC cohort | 794.73 (n = 40) | 733.26 (n = 42) | 92.27 | 81.27-104.7 | 44.6 |
| FDC cohort | 687.08 (n = 40) | 845.57 (n = 26) | 121.30 | 97.00-151.7 | 59.1 |
| Parameter | IV geometric LSM | Oral geometric LSM | LSM ratio (oral/IV) | 80% CI | Intrapatient CV% |
|---|---|---|---|---|---|
| Primary paired population | |||||
| 5-d AUClast, ng × hr per mL (primary end point) | |||||
| DC cohort (n = 40) | 802.81 | 750.82 | 93.52 | 82.10-106.5 | 47.0 |
| FDC cohort (n = 24) | 745.26 | 727.29 | 97.59 | 80.48-118.3 | 53.8 |
| Secondary unpaired population | |||||
| 5-d AUClast, ng × hr per mL | |||||
| DC cohort | 795.41 (n = 42) | 735.62 (n = 48) | 92.48 | 81.37-105.1 | 48.4 |
| FDC cohort | 742.26 (n = 26) | 760.43 (n = 28) | 102.45 | 85.35-123.0 | 52.7 |
| 5-d AUC24, h/ng per mL | |||||
| DC cohort | 794.73 (n = 40) | 753.68 (n = 45) | 94.83 | 83.97-107.1 | 43.5 |
| FDC cohort | 696.90 (n = 20) | 846.82 (n = 26) | 121.51 | 97.15-152.0 | 59.1 |
| 5-d AUC∞, ng × hr per mL | |||||
| DC cohort | 794.73 (n = 40) | 733.26 (n = 42) | 92.27 | 81.27-104.7 | 44.6 |
| FDC cohort | 687.08 (n = 40) | 845.57 (n = 26) | 121.30 | 97.00-151.7 | 59.1 |
AUC24, AUC from time 0 to 24 h; AUC∞, AUC from time 0 to ∞; CV, coefficient of variation; DC, dose confirmation (2 separate capsules of cedazuridine and decitabine); FDC, fixed dose combination.
Peak decitabine plasma concentrations were achieved at 1 hour following the start of the 1-hour IV infusion and also 1 hour after oral administration of oral cedazuridine/decitabine (Figure 2).

Mean decitabine plasma concentrations-time profiles following single and multiple oral doses of cedazuridine/decitabine, and following single IV infusion of decitabine during dose confirmation and fixed-dose combination stages. (A-B) Linear and (C-D) semilogarithmic plots are shown. LLOQ, lower limit of quantitation.
Mean decitabine plasma concentrations-time profiles following single and multiple oral doses of cedazuridine/decitabine, and following single IV infusion of decitabine during dose confirmation and fixed-dose combination stages. (A-B) Linear and (C-D) semilogarithmic plots are shown. LLOQ, lower limit of quantitation.
Pharmacodynamics
The treatment effect on LINE-1 demethylation was evaluated in 78 patients. In the ANOVA model, the absolute difference in maximum LSM %LINE-1 demethylation between oral and IV dosing was ∼1% or less with the 95% CI of the difference containing 0. There was no clinically or statistically significant difference in the effect on global DNA methylation between oral and IV dosing (Table 3).
Maximum %LINE-1 demethylation by treatment cycle
| Phase 2 overall stage cycle | Patients, n | Treatment | Mean baseline* | Maximum %LINE-1 demethylation change from baseline | Difference (oral–IV) in mean maximum %LINE-1 demethylation | ||
|---|---|---|---|---|---|---|---|
| LSM | 95% CI | Estimate | 95% CI† | ||||
| 1 | 40 | Oral C/D | 79.349 | 10.726 | 9.161-12.291 | −1.079 | −3.320 to 1.163 |
| 38 | IV decitabine | 79.119 | 11.805 | 10.200-13.410 | |||
| 2 | 31 | Oral C/D | 77.626 | 9.340 | 7.387-11.292 | −0.017 | −2.736 to 2.701 |
| 33 | IV decitabine | 77.298 | 9.357 | 7.465-11.249 | |||
| Phase 2 overall stage cycle | Patients, n | Treatment | Mean baseline* | Maximum %LINE-1 demethylation change from baseline | Difference (oral–IV) in mean maximum %LINE-1 demethylation | ||
|---|---|---|---|---|---|---|---|
| LSM | 95% CI | Estimate | 95% CI† | ||||
| 1 | 40 | Oral C/D | 79.349 | 10.726 | 9.161-12.291 | −1.079 | −3.320 to 1.163 |
| 38 | IV decitabine | 79.119 | 11.805 | 10.200-13.410 | |||
| 2 | 31 | Oral C/D | 77.626 | 9.340 | 7.387-11.292 | −0.017 | −2.736 to 2.701 |
| 33 | IV decitabine | 77.298 | 9.357 | 7.465-11.249 | |||
C/D, cedazuridine/decitabine.
Baseline for cycle 1 was last available value on or before day 1 of cycle 1; baseline for cycle 2 was value on day 1 of cycle 2.
Generated using ANOVA model separately for cycles 1 and 2.
Efficacy
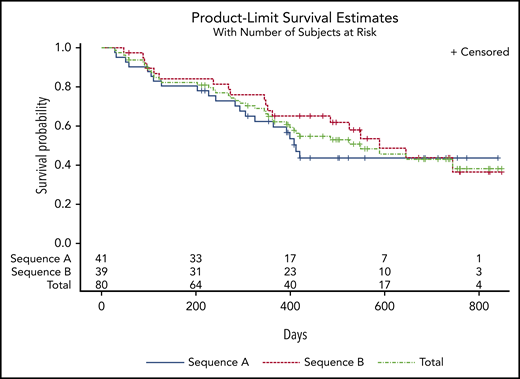
All treated patients were included in the efficacy analyses. Clinical responses were seen in 48 of 80 patients (60%), including 17 (21%) with complete responses (CR; Table 4). The median duration of CR was 13.3 months (95% CI, 6.5-13.8). Figure 3 shows time to first response and time to best response by cycle. Although response can manifest as early as the first cycle, most responses start to manifest by cycle 3, but best response can take up to 5 or more cycles. Of 38 patients who were red blood cell transfusion dependent at baseline, 19 (50%) became transfusion independent. Of the 12 patients who were platelet transfusion dependent at baseline, 6 (50%) became transfusion independent. Median time to AML or death for the overall population treated was 12.1 months (95% CI, 5.9-not estimable). Survival by randomized sequence and in the overall population is shown in Figure 4. Median overall survival for all patients treated was 18.3 months (95% CI, 9.1-not estimable).
Analysis of best response
| Type of response | Phase 2 overall (N = 80) | |
|---|---|---|
| n (%) | 95% CI | |
| CR | 17 (21) | 13-32 |
| PR | 0 | |
| mCR | 18 (22) | 14-33 |
| mCR with HI | 6 (7) | 3-16 |
| HI | 13 (16) | 9-26 |
| HI-E | 8 (10) | 4-19 |
| HI-N | 2 (2) | 0-9 |
| HI-P | 11 (14) | 7-23 |
| Overall response* (CR + PR + mCR + HI) | 48 (60) | 48-71 |
| No response | 32 (40) | 29-52 |
| Type of response | Phase 2 overall (N = 80) | |
|---|---|---|
| n (%) | 95% CI | |
| CR | 17 (21) | 13-32 |
| PR | 0 | |
| mCR | 18 (22) | 14-33 |
| mCR with HI | 6 (7) | 3-16 |
| HI | 13 (16) | 9-26 |
| HI-E | 8 (10) | 4-19 |
| HI-N | 2 (2) | 0-9 |
| HI-P | 11 (14) | 7-23 |
| Overall response* (CR + PR + mCR + HI) | 48 (60) | 48-71 |
| No response | 32 (40) | 29-52 |
CR, complete response; HI, hematologic improvement; HI-E, erythroid response; HI-N, neutrophil response; HI-P, platelet response; mCR, marrow complete response; PR, partial response.
Patients are counted only once with their best response as per the table hierarchy.

Time to first response and time to best response by cycle (N = 80). HI, hematologic improvement; mCR, marrow complete response.
Time to first response and time to best response by cycle (N = 80). HI, hematologic improvement; mCR, marrow complete response.
Safety
Table 5 summarizes the most common AEs (all grades and grade ≥3) regardless of causality in the overall treated population. The incidences of AEs during cycles 1 and 2 were similar between oral and IV treatment of all grades, and for grade ≥3. No notable increase in GI AEs was observed with oral decitabine vs IV decitabine in the 2 first randomized cycles.
Treatment-emergent AEs during cycles 1 and 2, and the entire phase 2 study regardless of relation to study treatment
| Preferred term, n (%) | IV decitabine cycle 1 or 2 (n = 75) | Oral cedazuridine/decitabine cycle 1 or 2 (n =78) | All oral cedazuridine/decitabine cycles (n = 78) |
|---|---|---|---|
| Patients with ≥1 TEAE | 69 (92) | 72 (92) | 75 (96) |
| Most common TEAEs (≥20% of patients) | |||
| Neutropenia | 22 (29) | 17 (22) | 36 (46) |
| Thrombocytopenia | 24 (32) | 23 (29) | 34 (44) |
| Fatigue | 10 (13) | 15 (19) | 26 (33) |
| Febrile neutropenia | 12 (16) | 9 (12) | 23 (29) |
| Nausea | 11 (15) | 13 (17) | 22 (28) |
| Diarrhea | 9 (12) | 10 (13) | 22 (28) |
| Leukopenia | 9 (12) | 10 (13) | 21 (27) |
| Dizziness | 8 (11) | 9 (12) | 20 (26) |
| Anemia | 11 (15) | 10 (13) | 19 (24) |
| Constipation | 12 (16) | 14 (18) | 19 (24) |
| Dyspnea | 2 (3) | 12 (15) | 19 (24) |
| Patients with grade ≥3 TEAEs | 44 (59) | 45 (58) | 65 (83) |
| Most common grade ≥3 TEAEs (≥10% of patients) | |||
| Neutropenia | 20 (27) | 16 (21) | 36 (46) |
| Thrombocytopenia | 21 (28) | 18 (23) | 30 (38) |
| Febrile neutropenia | 12 (16) | 9 (12) | 23 (29) |
| Leukopenia | 8 (11) | 7 (9) | 19 (24) |
| Anemia | 9 (12) | 9 (12) | 17 (22) |
| Pneumonia | 5 (7) | 7 (9) | 10 (13) |
| Sepsis | 1 (1) | 4 (5) | 8 (10) |
| Preferred term, n (%) | IV decitabine cycle 1 or 2 (n = 75) | Oral cedazuridine/decitabine cycle 1 or 2 (n =78) | All oral cedazuridine/decitabine cycles (n = 78) |
|---|---|---|---|
| Patients with ≥1 TEAE | 69 (92) | 72 (92) | 75 (96) |
| Most common TEAEs (≥20% of patients) | |||
| Neutropenia | 22 (29) | 17 (22) | 36 (46) |
| Thrombocytopenia | 24 (32) | 23 (29) | 34 (44) |
| Fatigue | 10 (13) | 15 (19) | 26 (33) |
| Febrile neutropenia | 12 (16) | 9 (12) | 23 (29) |
| Nausea | 11 (15) | 13 (17) | 22 (28) |
| Diarrhea | 9 (12) | 10 (13) | 22 (28) |
| Leukopenia | 9 (12) | 10 (13) | 21 (27) |
| Dizziness | 8 (11) | 9 (12) | 20 (26) |
| Anemia | 11 (15) | 10 (13) | 19 (24) |
| Constipation | 12 (16) | 14 (18) | 19 (24) |
| Dyspnea | 2 (3) | 12 (15) | 19 (24) |
| Patients with grade ≥3 TEAEs | 44 (59) | 45 (58) | 65 (83) |
| Most common grade ≥3 TEAEs (≥10% of patients) | |||
| Neutropenia | 20 (27) | 16 (21) | 36 (46) |
| Thrombocytopenia | 21 (28) | 18 (23) | 30 (38) |
| Febrile neutropenia | 12 (16) | 9 (12) | 23 (29) |
| Leukopenia | 8 (11) | 7 (9) | 19 (24) |
| Anemia | 9 (12) | 9 (12) | 17 (22) |
| Pneumonia | 5 (7) | 7 (9) | 10 (13) |
| Sepsis | 1 (1) | 4 (5) | 8 (10) |
TEAEs were coded using Medical Dictionary for Regulatory Activities, version 21.0, and are presented in decreasing incidence for entire phase 2 study; patients received IV decitabine or oral cedazuridine/decitabine in cycles 1 and 2, and then received oral cedazuridine/decitabine in all subsequent cycles.
TEAE, treatment-emergent adverse events.
Five patients discontinued treatment because of AEs, none of which were considered related to study therapy. Eleven patients had an AE with an outcome of death, including 4 from sepsis or septic shock and 2 from pneumonia (all considered not related to treatment), and 1 each from respiratory failure, cardiac arrest, sudden death, myocarditis, and small-cell lung cancer.
Discussion
This is the first phase 2 study to demonstrate that an oral HMA using a fixed dose of 35 mg of decitabine and 100 mg of the CDA inhibitor cedazuridine can achieve a similar systemic decitabine AUC exposure (93.5%-97.6%) compared with standard dose IV decitabine (20 mg/m2). The randomized crossover design allowed reliable intrapatient comparison in the first 2 cycles for PK, PD, and initial safety of the 2 treatments. The similar levels of LINE-1 demethylation noted between oral cedazuridine/decitabine and IV decitabine provide further evidence that the decitabine exposures achieved with oral dosing produced almost identical PD effects. These phase 2 data confirm the findings of the phase 1 dose-escalation study in which cedazuridine 100 mg plus decitabine 30 or 40 mg yielded 5-day AUC exposures that were 81% and 128% of IV decitabine, respectively.20 The incidences of AEs in the first 2 randomized cycles regardless of causality (all grades and grade ≥3) were similar between oral and IV treatment, with no appreciable differences in frequency or severity. Notably, GI AEs were predominantly grade 1 or 2 and were reported at similar incidences between oral and IV dosing in cycles 1 and 2, suggesting no additional GI toxicity with oral treatment, at least initially during the first 2 cycles.
Evidence of clinical activity was observed in the phase 2 study, with 21% of patients achieving a best response of CR with a median duration of 13.3 months. Overall, 60% of patients had a clinical response. Red blood cell and platelet transfusion independence was achieved in 50% of patients who were transfusion dependent at baseline. These efficacy data compare well with those previously reported with 5-day IV decitabine by Steensma et al13 : 17% CR, 51% overall response, and 32% to 40% transfusion independence rates. Time to best response may take up to 5 or more cycles, which is consistent with data reported by Steensma et al,13 and supportive of epigenetic DNA methylation inhibition as the important mechanism of action.4 Median overall survival of 18.3 months also compares well with the 19.4 months reported by Steensma13 and the 15 to 24.5 months reported with the single agent azacitidine (administered IV or subcutaneously) in a review of such studies by Zeidan et al.23
Recently, CC-486, an oral azacitidine formulation with a distinct PK/PD profile from injectable azacitidine, demonstrated significant survival benefit compared with placebo as maintenance treatment after intensive chemotherapy in patients with AML who were not eligible for hematopoietic stem cell transplant.24 CC-486 was given at a high dose of 300 mg/d over an extended schedule of 14 days every 28 days. Oral cedazuridine/decitabine has a similar decitabine PK/PD profile to IV decitabine, and thus is the only oral HMA with similar systemic exposure to its injectable form using a low dose of decitabine (35 mg) and the same 5-day schedule.
The main limitations of this study were the lack of safety and efficacy comparisons between treatments over the entire phase 2 treatment courses because the randomization between oral and IV was only for the first 2 cycles. A parallel-arm, randomized design between oral and IV dosing for the entire treatment courses would require a prohibitively large number of patients to provide sufficient power to show noninferiority based on clinical end points. Such a design would also be difficult to enroll because patients would probably have a preference to receive oral treatment. Given the similar decitabine AUC exposures, DNA demethylation, and safety in the first 2 randomized cycles of oral vs IV treatment, and the clinical response data that are consistent with what has previously been reported for IV decitabine, we believe such a clinical study is not warranted. Another limitation of the study is the relatively small number of patients treated in the FDC cohort. For that reason, a larger study of ∼130 patients is under way, with a similar design to establish systemic oral/IV decitabine AUC equivalence (90% CI, 0.8-1.25) using the oral FDC tablet of cedazuridine/decitabine and IV decitabine (Study of ASTX727 vs IV Decitabine in MDS, CMML, and AML; www.clinicaltrials.gov, #NCT03306264).
Taken together, these findings demonstrate that the equivalent decitabine AUC exposures after oral treatment (at the fixed doses of cedazuridine 100 mg and decitabine 35 mg) result in a clinical efficacy and safety profile consistent with the profile previously reported with standard IV decitabine dose of 20 mg/m2. The availability of an oral form of decitabine with the same decitabine exposure as IV offers promising opportunities in the treatment of myeloid malignancies. In addition to its established use in MDS and CMML, the benefit of IV decitabine also expands to patients with AML who are deemed not to be candidates for intensive chemotherapy either as a single agent10 or in combination with venetoclax.25 Furthermore, the availability of oral decitabine offers attractive opportunities for future development of oral combinations with the hope of further improving treatment outcomes and potentially enhancing quality of life.
Presented in abstract form at the 22nd Congress of the European Hematology Association, Madrid, Spain, 22-25 June 2017; the 59th American Society of Hematology Annual Meeting, Atlanta, GA, 9-12 December 2017, Atlanta, GA; and the 15th International Symposium on MDS, Copenhagen, Denmark, 5-8 May 2019.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
For original data, please e-mail the corresponding author.
Acknowledgments
This trial was supported by Astex Pharmaceuticals, Inc. Geoff Marx and Barry M. Weichman of BioScience Communications, New York, NY (a medical communications company), provided medical writing and editorial assistance to the authors during preparation of this manuscript, and were funded by Astex.
Authorship
Contribution: G.G.-M., A.O., M.A., and M.R.S. conceived and designed the study; all authors except A.O. and M.A. enrolled patients and contributed data; G.G.-M., E.A.G., D.P.S., A.O., M.A., and M.R.S. interpreted data and wrote the manuscript; and all authors reviewed the manuscript and approved the final version.
Conflict-of-interest disclosure: G.G.-M. has received research funding and honoraria from Astex/Otsuka. E.A.G. has consulted for Astex, AbbVie, Boston Scientific, Celgene, New Link Genetics, Novartis, Otsuka, Palmer, and Persimmune; has received research funding from Astex, Celgene, Genentech, and Otsuka; and has served as Principal Investigator on clinical trials for Appelis and Onconova. D.P.S. has received research funding from Celgene, H3 Biosciences, and Janssen; and has received personal fees from Janssen, Onconova, Sensei, and Takeda. G.J.R. has consulted or served on advisory boards or data and safety monitoring committees for Astex, AbbVie, Actinium, Agios, Amphivena, Argenx, Astellas, Bayer, Celgene, Celltrion, Daiichi Sankyo, Eisai, Janssen, Jazz, MEI, Novartis, Orsenix, Otsuka, Pfizer, Roche/Genentech, Sandoz, Takeda, and Trovagene; and has received research support from Cellectis. R.W. has received honoraria and research funding from Alexion, Celgene, and Novartis. O.O. has received research funding from Astex. AbbVie, Agios, AstraZeneca, Celgene, CTI/Baxalta, Gilead, Incyte, Janssen, NS-Pharma, Oncotherapy, Sanofi, and S*Bio; has served on advisory boards for AbbVie, Celgene, CTI/Baxalta, Dava Oncology, Incyte, Jazz, and Pfizer; and has received drug supply support from Pfizer. K.Y. has received research funding from Astex, MedImmune, Millennium, MSD, and Roche/Genentech; has received honoraria from Novartis and Pfizer; and has served on boards of directors or advisory committees for Astellas, Celgene, Novartis, Pfizer, and Takeda. L.B. has consulted for BMS, Novartis, Paladin, and Pfizer; and has received royalties for patents from ExCellThera. C.O. has served on boards of directors or advisory committees for Astex, BMS, Pfizer, and Shionogi; and has received research funding from Astex and Genentech. L.C.M. has received research funding from Jazz; has consulted for Incyte; has served on a speakers bureau for Celgene; has served on advisory boards for Novartis and TG Therapeutics; and has equity in Pfizer. J.B. has consulted for and received honoraria from Celgene, Jazz, Novartis, and Pfizer; has received honoraria from Otsuka; and has received research funding from Celgene, Pfizer, and Roche. H.K. has received research funding from Astex, AbbVie, Agios, Amgen, Ariad, BMS, Cyclacel, Immunogen, Jazz, and Pfizer; has received honoraria from AbbVie, Agios, Amgen, Immunogen, Orsinex, Pfizer, and Takeda; and has served on advisory boards for Actinium. A.O. and M.A. are employees of Astex. M.R.S. has received research funding from Astex, Incyte, Millennium, and TG Therapeutics; has consulted for Astex, Celgene, BMS, Incyte, Karyopharm, Millennium, Ryvu, Sierra Oncology, and TG Therapeutics; and has equity in Karyopharm and a licensed patent from Boehringer Ingelheim. The remaining authors declare no competing financial interests.
Correspondence: Guillermo Garcia-Manero, Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd-428, Houston, TX 77030-4009; e-mail: ggarciam@mdanderson.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal