

Key Points
A VTE case/control exome sequencing study identified rare variants in the genes encoding protein S, protein C, antithrombin, and stabilin-2.
Rare damaging variants in STAB2 are associated with increased plasma von Willebrand factor through decreased clearance.

Abstract
Deep vein thrombosis and pulmonary embolism, collectively defined as venous thromboembolism (VTE), are the third leading cause of cardiovascular death in the United States. Common genetic variants conferring increased varying degrees of VTE risk have been identified by genome-wide association studies (GWAS). Rare mutations in the anticoagulant genes PROC, PROS1 and SERPINC1 result in perinatal lethal thrombosis in homozygotes and markedly increased VTE risk in heterozygotes. However, currently described VTE variants account for an insufficient portion of risk to be routinely used for clinical decision making. To identify new rare VTE risk variants, we performed a whole-exome study of 393 individuals with unprovoked VTE and 6114 controls. This study identified 4 genes harboring an excess number of rare damaging variants in patients with VTE: PROS1, STAB2, PROC, and SERPINC1. At STAB2, 7.8% of VTE cases and 2.4% of controls had a qualifying rare variant. In cell culture, VTE-associated variants of STAB2 had a reduced surface expression compared with reference STAB2. Common variants in STAB2 have been previously associated with plasma von Willebrand factor and coagulation factor VIII levels in GWAS, suggesting that haploinsufficiency of stabilin-2 may increase VTE risk through elevated levels of these procoagulants. In an independent cohort, we found higher von Willebrand factor levels and equivalent propeptide levels in individuals with rare STAB2 variants compared with controls. Taken together, this study demonstrates the utility of gene-based collapsing analyses to identify loci harboring an excess of rare variants with functional connections to a complex thrombotic disease.
Introduction
Venous thromboembolism (VTE), including deep vein thrombosis and pulmonary embolism, is a complex genetic trait determined by multiple genetic and environmental influences. Prolonged hospitalization, surgery, and immobilization are the strongest risk factors for VTE as well as advancing age, estrogen therapy, cancer, chemotherapy, obesity, pregnancy, and smoking.1 Genetic factors play a major role in VTE risk, especially in patients with unprovoked VTE (without an identified environmental risk factor).2 A recent sibling study estimated the heritability of VTE at 40% for males and 47% for females.3 Several individual genome-wide association studies (GWAS) and metaanalyses for VTE have been reported,4-13 identifying signals at loci containing common gene variants already known to be associated with VTE, including the ABO blood type, factor V Leiden, and prothrombin 20210. Of note, these studies also provide convincing evidence to exclude any significant contribution from several other previously implicated common gene variants, including the SERPINE1 4G/5G and MTHFR C677Tpolymorphisms.14 GWAS based on single-nucleotide polymorphism (SNP) arrays and genotype imputation are generally unable to capture rare variant genotypes and can be insufficiently powered to detect the effect of genetic variants with minor allele frequency <1%. A recent analysis using whole-genome sequencing data suggests that such rare genetic variants account for most of the “missing heritability” for human height not captured by GWAS, supporting the investigation of rare variants in other common complex traits.15
Autosomal dominant forms of thrombophilia have been identified in families, generally explained by rare loss of function mutations in one of the 3 natural anticoagulant proteins: protein C,16,17 protein S,18,19 or antithrombin.20,21 An excess of rare damaging variants in these 3 genes was recently reported in 68 individuals with out-of-hospital fatal pulmonary embolism.22 Exome sequencing of patients with VTE is beginning to be used in clinical settings emphasizing the need for variant discovery studies.23,24
We now report results from a gene-based collapsing analysis of whole-exome sequencing data from 393 individuals with VTE compared with 6114 controls. We identified an excess of damaging variants among VTE patients for the genes encoding the anticoagulants protein C, protein S, and antithrombin, as well as STAB2, a gene encoding an endothelial scavenger receptor. We examined selected VTE STAB2 variants in a cell culture model and found altered intracellular trafficking and reduced cell surface expression compared with reference STAB2. We also studied individuals from an independent cohort and found elevated plasma von Willebrand factor (VWF) levels in individuals harboring rare variants in STAB2.
Patients and methods
Case and control samples for exome sequencing
Deidentified DNA samples from individuals with VTE were collected as part of 3 previous clinical and genetic studies of VTE. The study was approved by the University of Michigan institutional review board and conducted in accordance with the Declaration of Helsinki. One set of samples was derived from the ELATE study, which was designed to compare the effectiveness of different warfarin regimens after unprovoked VTE.25,26 Of the 661 DNA samples available from ELATE, 135 were selected for exome sequencing. The second set of samples was derived from the DODS study, which was designed to analyze the effectiveness of plasma d-dimer as a biomarker for recurrent VTE.27,28 Of the 219 DNA samples available, 97 were selected for exome sequencing. Samples from ELATE and DODS were selected based only on quality of DNA and without knowledge of age, ethnicity, sex, or severity of clinical disease. The third set of samples was taken from the GIFT study, an affected sibling study of VTE.29,30 In GIFT, we sequenced 1 affected proband from each family for a total of 201 samples. Further demographic information on the participants of the ELATE, DODS, and GIFT studies are available in the original referenced studies. Controls were sourced from the Institute for Genomic Medicine (IGM) at Columbia University. Samples were selected for inclusion as controls if previously approved for control use and either whole genome sequenced or sequenced using a Nimblegen EZCap V5 (Roche-Nimblegen, Madison, WI) or Agilent 65MB exome kit (Agilent, Santa Clara, CA). In addition, samples were required to have passed IGM automated sample level QC with regards to sequencing quality, sex concordance, and cryptic relatedness and were of self-declared Caucasian ethnicity. The controls were not screened for a history of VTE.
Exome sequencing
DNA libraries were prepared at the University of Michigan DNA sequencing core (N = 296) using the Agilent SureSelect (n = 8; 2012) or Nimblegen V3 (n = 288; 2014) systems to enrich for protein coding genomic DNA. Additional samples from the GIFT study were sequenced at Marseilles (N = 96; 2013) using the Illumina TruSeq to enrich for exonic DNA. FASTQ files from both centers were transferred to the IGM at Columbia University, where sequence data from 6114 control exomes were available. Further details on the variant calling pipeline, sample selection, and gene-based collapsing analysis are available in the supplemental data, available on the Blood Web site.
TwinsUK analysis
Plasma from 4000 participants (2000 sibling pairs) in the TwinsUK study was characterized using VWF and VWF propeptide (VWFpp) AlphaLISA assays as previously described.31 Levels were mean centered, log transformed, and adjusted for ABO genotypes. Whole-genome sequencing data were available for 1162 of the 4000 plasmas. VWF and VWFpp levels were compared between individuals with and without qualifying variants in STAB2 using the Mann-Whitney U test implemented in Prism 8 (GraphPad Software, San Diego, CA).
Stabillin-2 characterization in vitro
Further details on the cloning and creation of stable cell lines expressing reference and STAB2 missense stabilin-2 can be found in the supplemental data. Primers used to generate and sequence these plasmid constructs are listed in supplemental Table 1. To limit the variation of STAB2 expression due to insertion site, complementary DNA (cDNA) encoding reference STAB2 or selected missense variants was inserted into a specific locus in TRex 293 cells using flp recombinase. We characterized the relative expression of STAB2 messenger RNA using reverse transcription polymerase chain reaction with TaqMan probes specific for STAB2, TOP1, and GAPDH (supplemental data; supplemental Figure 1). Synthesis of recombinant stabilin-2 protein in these cell lines was confirmed by western blotting and confocal microscopy (supplemental data; supplemental Figure 2). Stable cell lines were maintained with 5 μg/mL of blasticidin.
Flow cytometry
Cells were acquired and analyzed using an LSR II flow cytometer (BD Biosciences, San Jose, CA) and FlowJo software (FlowJo, Ashland, OR). Cells were fixed in 4% formaldehyde in phosphate-buffered saline (PBS) for 15 minutes, washed 3 additional times, and resuspended in PBS for subsequent analysis via flow cytometry. Antibodies used in flow cytometry analysis were rabbit anti-human stabilin-2 c-terminal domain (Sigma-Aldrich) and mouse anti-stabilin-2 ectodomain (courtesy of Edward Harris, MAb30). Secondary antibodies were goat anti-mouse immunoglobulin G (IgG) AlexaFluor 488 (Invitrogen) and goat anti-rabbit IgG AlexaFluor 647 (Invitrogen).
On-Cell Western
Cultured stable cells and untransfected cells Flp-In T-REx (Invitrogen) were grown in poly-d-lysine (Millipore-Sigma) treated 24-well tissue culture plates (Fisher Scientific) overnight and then induced with 1 ug/mL tetracycline (Sigma) for an additional 24 hours until they reached 70% to 90% confluency. Cells were then blocked with 1× Dulbecco's PBS (Gibco)/1% bovine serum albumin (Sigma)/2% normal donkey serum (Jackson Immunoresearch) and incubated with anti-STAB2 (courtesy of Edward Harris, MAb154) primary antibody and then washed and incubated with a donkey anti-mouse IgG (IRDye 800CW, LI-COR). Cells were washed with Dulbecco's PBS, and DRAQ5 cell stain (Thermo Scientific) was added for a 5-minute incubation. Cells were then imaged, and signals were quantified on LI-COR Odyssey CLx infrared imaging system (LI-COR).
Results
Gene collapsing analysis of VTE
Whole-exome sequencing was performed on 393 DNA samples from 3 different clinical studies of VTE as described above. After selection of samples that were unrelated and of European ancestry (determined by principal component analysis of genotypes), 373 VTE case exomes were available for comparison with 5784 control samples. The mean (standard deviation) percent of CCDSr14 bases covered 10× in cases was 0.9598 (0.0169) and in controls was 0.9615 (0.0162). The difference between cases and controls in coverage was not significant (Wilcox test, P = .3620913). The factor V Leiden variant (rs6025) was present in 18.4% of our cases and 2.3% of controls, consistent with prior studies showing similar allele frequencies in European VTE case/control studies.5,8 Nonsense, frameshift, splice site, or nonbenign (PolyPhen2; supplemental Figure 3) missense single nucleotide variants were included. Only variants with allele frequencies <0.05% across EVS, ExAC global, and subpopulations, as well as within the internal case/control dataset, were selected for the collapsing analysis because this low frequency better captured rare coding variation with large odds ratios (ORs; supplemental Figure 4). The dominant genetic model corresponding with the known inheritance patterns for familial thrombophilia due to antithrombin, protein C, or protein S deficiency was used. We did not include common ABO blood type, factor V Leiden, or prothrombin 20210 defining SNPs in our collapsing analyses because their contribution to VTE risk is well documented, and they are present in European populations well above allele frequency thresholds used in our collapsing analysis.
Exome sequencing identified a total of 430 870 qualifying variants in cases and controls. The gene-based collapsing analysis included 18 669 CCDS annotated genes. We thus set the exome-wide significance threshold at Bonferroni corrected P value .05/18 669 = 2.6 × 10−6. The top 4 genes by P value were PROS1 (P = 2.01 × 10−9, OR 11.8), STAB2 (P = 2.70 × 10−7, OR 3.37), PROC (P = 3.24 × 10−5, OR 11.0), and SERPINC1 (P = 1.10 × 10−4, OR 8.49; Figure 1A). The quantile-quantile plot (Figure 1B), demonstrated good control of systematic bias and deviation from null hypothesis only at the lowest P values. Case/control differential burden did not meet exome-wide significance in PROC or SERPINC1, although these genes’ well-characterized roles in the anticoagulation pathway suggest that they may in fact represent true signal that lacked adequate statistical power to formally implicate. A list of the top 25 genes in the collapsing analysis and the list of annotated variants identified at the top 4 loci in cases and controls can be found in supplemental Tables 2-6.
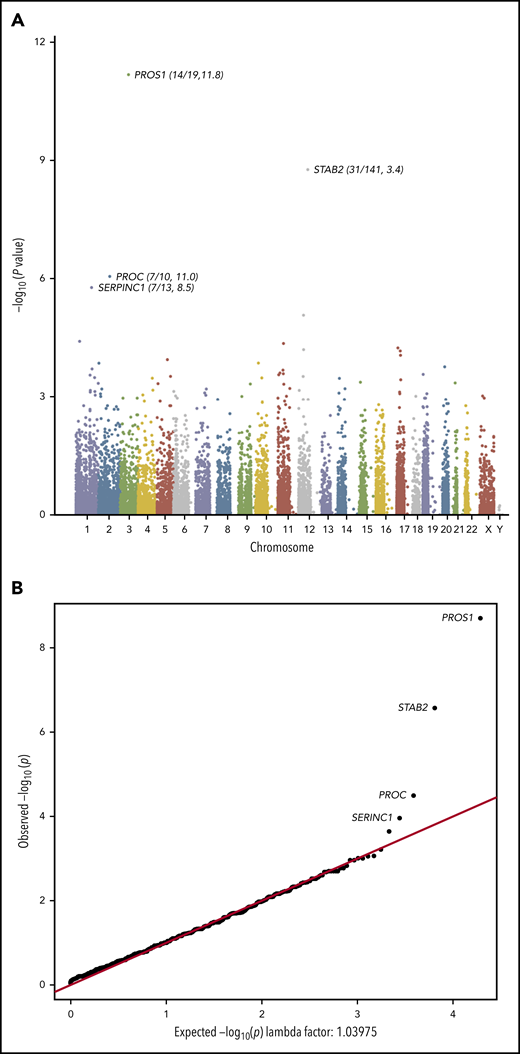
Collapsing test results from exome sequencing. We tested the coding sequence of 17 862 genes for greater than expected number of rare damaging variants in 373 cases vs 5784 controls. Gene names are indicated for top 4 signals. (A) Manhattan plot of −log10(p) values for each gene by Fisher’s exact test. Each dot represents one of 17 862 tested genes. Number of variants and OR (cases/controls, OR) indicated for top 4 genes. (B) Quantile-Quantile plot of observed −log10(p) (y-axis) vs expected (x-axis) by Fisher’s exact test. Each dot represents one of 17 862 tested genes.
Collapsing test results from exome sequencing. We tested the coding sequence of 17 862 genes for greater than expected number of rare damaging variants in 373 cases vs 5784 controls. Gene names are indicated for top 4 signals. (A) Manhattan plot of −log10(p) values for each gene by Fisher’s exact test. Each dot represents one of 17 862 tested genes. Number of variants and OR (cases/controls, OR) indicated for top 4 genes. (B) Quantile-Quantile plot of observed −log10(p) (y-axis) vs expected (x-axis) by Fisher’s exact test. Each dot represents one of 17 862 tested genes.
The strongest signal in the collapsing analysis was generated by variants in the PROS1 locus. Here, 14 of 373 cases had qualifying variants (3.7%), whereas only 19 of 5784 controls had a qualifying variant (0.3%). Most case variants in PROS1 were missense, although 1 nonsense variant was identified. All PROS1 variants identified in controls were missense.
At the STAB2 locus genotype level, our study identified 29 distinct qualifying variants in 373 case exomes (7.8%) compared with 106 variants in 5784 control exomes (1.8%). This corresponded to 29 case individuals and 141 controls with at least 1 qualifying variant in STAB2. Three case variants were found in an additional case, and 8 case variants were also found in controls. Three cases had compound heterozygous qualifying variants in STAB2. The proportion of STAB2 variants that were loss of function (stop gained, frameshift, splice donor/acceptor) was significantly greater in cases than controls (8 of 29 qualified cases vs 15 of 141 controls, OR 3.17, P = .03). The collapsing statistics for only loss-of-function variants in STAB2 are consistent with this finding (8 of 373 cases vs 15 of 5784 controls, 2.0%/0.3%, OR 8.4, P = 3.7 × 10−5). Three of 29 (9.7%) individuals with qualifying STAB2 variants were also heterozygous for factor V Leiden, which was lower than all cases (18.5%).
In order to compare case/control burden while taking into account the potential mixture of effect sizes and directions exerted by rare coding variation, we used the SKAT-O method to weight variants by their minor allele frequency. In supplemental Figure 5, the results of SKAT-O testing are displayed. Here, PROC, STAB2, and PROS1 had exome-wide significant signals (P = 5e-9, 6e-7, and 8e-7, respectively), whereas SERPINC1 was now ranked 11 in the full top results (P = 4e-4). No other genes had exome-wide significance.
Focus on STAB2 variants in VTE cases
The STAB2 locus on chromosome 12 has 69 exons >7659 bp and encodes stabilin-2, a class H scavenger receptor expressed predominantly in the sinusoidal endothelium of the liver and spleen.32 Stabilin-2 is a single transmembrane domain glycoprotein with previously described roles in the clearance from circulation of a variety of plasma ligands, including heparan sulfate, keratin sulfate, hyaluronic acid, and chondroitin sulfate.33,34 In recent GWAS, variants in STAB2 and several other loci have been associated with variation in plasma VWF levels.11,35,36 VWF is a central mediator of hemostasis, facilitating platelet binding to areas of endothelial injury and serving as a critical cofactor for circulating coagulation factor VIII. Importantly, elevations in plasma VWF are a well-described risk factor for VTE.37 Endothelial cells expressing stabilin-2 have recently been reported to bind and internalize VWF in vitro, whereas mice with engineered Stab2 deficiency exhibit a prolonged half-life of infused human plasma–derived VWF compared with controls, suggesting a possible direct connection between stabilin-2 function and plasma VWF concentration.38
In addition to an X-linked c-type lectin-like domain, stabillin-2 has multiple fasciclin-like and epidermal growth factor–like domain motifs, but detailed structure function information on these domains in stabilin-2 are lacking.34,39 We identified mutations in STAB2 across the entire coding sequence in both cases and controls without an overt clustering to a specific sequence-defined functional domain (Figure 2). Although nonsense, splice site, and frameshift variants strongly suggest loss of function, the functional consequences of missense variants are more difficult to predict without functional testing. Therefore, we further characterized selected case missense variants in a cell culture model.

Location of STAB2 variants in mature protein. (A) 373 cases and (B) 5784 controls. Protein map generated (modified) for STAB2 using Mutation Mapper (http://www.cbioportal.org/mutation_mapper). Annotated domains are labeled and indicated in the legend. Height of each lollipop indicates the number of qualifying variants at each position in stabilin-2. EGF, epidermal growth factor like.
Location of STAB2 variants in mature protein. (A) 373 cases and (B) 5784 controls. Protein map generated (modified) for STAB2 using Mutation Mapper (http://www.cbioportal.org/mutation_mapper). Annotated domains are labeled and indicated in the legend. Height of each lollipop indicates the number of qualifying variants at each position in stabilin-2. EGF, epidermal growth factor like.
Missense variants have reduced surface expression of stabilin-2
We selected 7 missense variants for further study based upon their inheritance pattern and their frequency in VTE cases but not on specific amino acid substitutions or amino acid residue location. In doing so, we attempted to select variants that potentially had either strongly negative effects on stabilin-2 function or potentially minimal impact on stabilin-2 function. Variants G1597S and I2124T were compound heterozygous (unknown phase) with a nonsense mutation, R243* and E2123*, respectively, which would reduce the “need” for those variants to be damaging in an autosomal dominant model. G1565S and G1592R were compound heterozygous in the same individual, making it unclear which variants were driving risk. G785E, E1669Q, and G2018R were all identified in 2 individuals with VTE, suggesting that their overall allele frequencies are higher or that they occurred more frequently in cases for a functional reason.
To determine cell surface expression differences between reference STAB2 and VTE case variants, we quantified stabilin-2 protein on the plasma membrane using the On-Cell Western technique.40 In 5 replicate assays using 2 independent clones for each variant, reference sequence stabilin-2 had significantly more surface expression signal compared with all other tested variants (Figure 3A; P < .0001, except G1597S, P = .0014, 2-tailed Student t test).
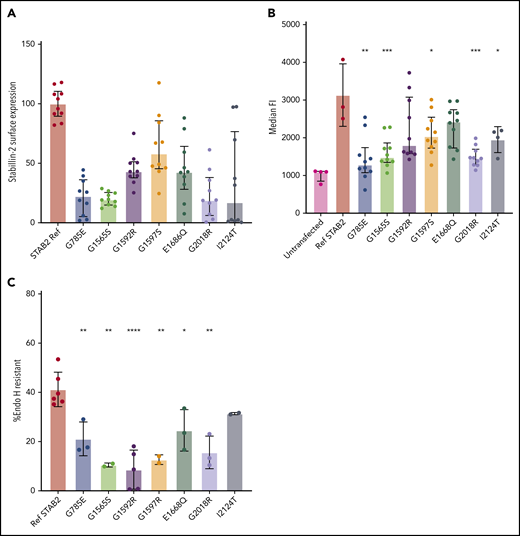
Analysis of selected STAB2 missense mutants. Reference STAB2 cDNA or cDNA encoding 7 different missense variants was used to establish stable stabilin-2 expressing cell lines in flp in TREx 293 cells. (A) On-Cell Western quantification of stabilin-2 surface expression was performed using an anti-ectodomain antibody and quantified with a Li-Cor Odyssey CLx and Image Studio software. IR antibody signal was normalized to cell counts, and reference sequence was mean centered to 100. All VTE variants had lower mean surface expression (P < .0001, except G1597S, P = .0014, 2-tailed Student t test). (B) Flow cytometry on live cells using an antibody against the receptor ectodomain to estimate median fluorescent signal corresponding to the amount of stabilin-2 receptor on the surface of the cells. (C) Cell lysate was obtained and incubated with and without Endo H and then western blotted using fluorescently conjugated secondary antibodies. Bands were quantified using image studio software, and the ratio of undigested to total bands was plotted for each mutant. For panels B and C, P values are reported in comparison of missense variant to ref STAB2. *P < .05, **P < .005, ***P < .0005, ****P < .0001 by 2-tailed Student t test.
Analysis of selected STAB2 missense mutants. Reference STAB2 cDNA or cDNA encoding 7 different missense variants was used to establish stable stabilin-2 expressing cell lines in flp in TREx 293 cells. (A) On-Cell Western quantification of stabilin-2 surface expression was performed using an anti-ectodomain antibody and quantified with a Li-Cor Odyssey CLx and Image Studio software. IR antibody signal was normalized to cell counts, and reference sequence was mean centered to 100. All VTE variants had lower mean surface expression (P < .0001, except G1597S, P = .0014, 2-tailed Student t test). (B) Flow cytometry on live cells using an antibody against the receptor ectodomain to estimate median fluorescent signal corresponding to the amount of stabilin-2 receptor on the surface of the cells. (C) Cell lysate was obtained and incubated with and without Endo H and then western blotted using fluorescently conjugated secondary antibodies. Bands were quantified using image studio software, and the ratio of undigested to total bands was plotted for each mutant. For panels B and C, P values are reported in comparison of missense variant to ref STAB2. *P < .05, **P < .005, ***P < .0005, ****P < .0001 by 2-tailed Student t test.
To further quantify the surface expression of stabilin-2 protein, we performed flow cytometry using antibodies specific to the stabilin-2 ectodomain. Reference stabilin-2 exhibited the highest surface expression signal compared with cell lines expressing missense variants, although variants G1592R and E1668Q did not reach a statistically significant difference compared with reference stabilin-2 (Figure 3B; P = .134, .066, respectively, 2-tailed Student t test).
In order to estimate the proportion of stabilin-2 receptor exiting the endoplasmic reticulum and entering the Golgi, cell lysates containing stabilin-2 protein were collected, denatured, and digested with Endo H.39 Western blots revealed protein bands from digested and protected forms of stabilin-2 for all variants (supplemental Figure 6), and % Endo H resistance was calculated (Figure 3C). Reference stabilin-2 protein had the highest proportion of Endo H resistance, consistent with more efficient exit from the ER and transport to the Golgi37,38 compared with case variants. Of the stabilin-2 variants, only I2124T (a variant detected in an individual with an additional variant E2123*) was equivalent to reference protein (P = .1086, 2-tailed Student t test).
Plasma VWF levels are elevated in individuals with rare STAB2 variants
If stabilin-2 functions as a major clearance receptor for VWF in humans, we hypothesized that individuals with damaging variants in STAB2 would have elevated plasma VWF levels due to a longer half-life in circulation. We measured plasma VWF levels in 4000 twin pairs from the TwinsUK study.41 We also measured VWFpp levels as a way to in order to determine if the change in VWF levels was due to altered clearance.31,42 We identified 1162 individuals who had whole-genome sequencing available43 and identified 38 individuals (3.2%) with 1 qualifying variant in STAB2 as defined by allele frequency and PolyPhen criteria used in our VTE case control study. This was similar in frequency to the controls (2.4%) examined in our primary collapsing analysis. VWF and VWFpp levels were log transformed to normalize distributions and adjusted for height, weight, and ABO status (determined by genotyping),44 leaving 31 individuals with and 996 without a qualifying variant in STAB2 (Figure 4). The STAB2−/+ group had higher VWF levels than the STAB2+/+ group (P = .0099, Mann-Whitney U test) but equivalent VWFpp levels (P = .9320, Mann-Whitney U test), suggesting that the difference in VWF levels was due to altered clearance
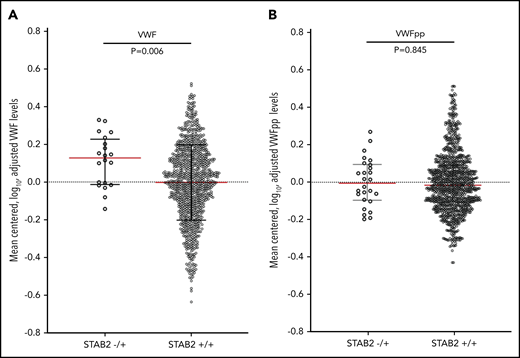
VWF and VWFpp levels were measured in plasma from the TwinsUK study. (A) VWF and (B) VWFpp levels were mean centered, log transformed, and adjusted for height, weight, and ABO blood type status predicted by genotype. STAB2−/+ indicated individuals with 1 qualifying variant in STAB2 (N = 31), while STAB2+/+ indicates individuals with no qualifying variant (n = 996). Black lines indicate interquartile range, and red line indicates median. P values for the difference in the distributions between STAB2−/+ and +/+ individuals were calculated using the Mann-Whitney U test.
VWF and VWFpp levels were measured in plasma from the TwinsUK study. (A) VWF and (B) VWFpp levels were mean centered, log transformed, and adjusted for height, weight, and ABO blood type status predicted by genotype. STAB2−/+ indicated individuals with 1 qualifying variant in STAB2 (N = 31), while STAB2+/+ indicates individuals with no qualifying variant (n = 996). Black lines indicate interquartile range, and red line indicates median. P values for the difference in the distributions between STAB2−/+ and +/+ individuals were calculated using the Mann-Whitney U test.
Discussion
Although several well-powered VTE GWAS have identified common genetic variant associations, they do not fully account for the heritable risk for this complex disease. In the present study, we uncovered excess mutation burden in the 3 major anticoagulant genes as well as a statistically significant association with a new candidate gene for VTE risk, STAB2. For a genomic study of a complex genetic trait, this study was limited by the relatively small number of case individuals who had exome sequencing data available. In addition, the imbalance between the number of cases and controls could have led to signal inflation. However, limiting our analysis to individuals of European ancestry, variants of very low frequencies and identical variant calling pipelines for case and control exomes generated a QQ plot that suggests that signal inflation was well controlled. By including only variants with very low allele frequencies, we may have missed contributions to VTE risk by variants with allele frequencies that were too low to be included in GWAS or accurately imputed in other genotype-based association studies but higher than our studies’ allele frequency limit. The collapsing analysis method increases power to detect rare variant associations and has been used to study other complex traits45 but is limited by the assumption that all qualifying variants have equal likelihood of altering protein function in the same effect direction. Indeed, this method ignores the possibility that some missense variants could lead to gain-of-function mutations or that some qualifying variants have no detrimental effect. In order to compare case/control burden while taking into account the potential mixture of effect sizes and directions exerted by rare coding variation, we used the SKAT-O method to weight variants by their minor allele frequency but did not identify significantly different results. In addition, lack of interrogation of the noncoding sequence in our study could not detect the effect of noncoding regulatory variants. Despite these limitations, 3 of the top 4 signals have previously been validated, suggesting that the clinical phenotyping of these cases was excellent and the overall rare variant architecture of VTE is relatively oligogenic, being determined by a limited number of higher effect size genes.
ORs calculated for the 3 anticoagulant gene associations were as high as expected, agreeing with previous studies in familial VTE.16-21 These high ORs are consistent with a pattern observed in many genomic studies where variants with very low allele frequencies are associated with larger effect sizes than more common variants.46 The OR for STAB2 in our collapsing analysis was lower than any of the 3 natural anticoagulant genes and more comparable to those estimated for more common SNPs encoding factor V Leiden and the prothrombin 20210 variants.5 However, when we include only loss-of-function variants (nonsense, splice site, and frameshift) for STAB2 in our collapsing analysis, the OR increases to 8.4, which is more comparable to the natural anticoagulant deficiencies. This finding emphasizes the need for a more comprehensive functional analysis of rare missense variants in STAB2 to understand which qualifying missense variants are functional in order to calculate a more accurate OR. Examination of the gnomAD database revealed a greater tolerance for loss of function variants in STAB2 compared with the PROC, PROS1, and SERPINC1 genes with a loss of function observed to expected (90% CI) ratio of 0.76 (0.65 to 0.88) compared with PROS1, PROC, and SERPINC1, which had ratios of 0.35 (0.23 to 0.57), 0.52 (0.33 to 0.86), and 0.0 (0.0 to 0.15), respectively.47
Genetic determinants of VTE alter the balance of procoagulant and anticoagulant forces to favor thrombosis. Likewise, genetic factors associated with altered plasma levels of important blood clotting proteins have been detected in VTE GWAS.11 As elevated plasma VWF levels have been linked to increased VTE risk, we expect that common variants associated with increased VWF would also be detected in GWAS for VTE. This is readily apparent with strong GWAS signals at the ABO locus for both VTE and VWF. On the other hand, it is unclear if loci from large GWAS studies will also harbor rare variants associated with the same complex genetic trait. Low-frequency variants in STAB2 are associated higher VWF levels,35,36 rs141041254 (p.2377K), but with the exception of subthreshold associations with common STAB2 SNPs,4,13 STAB2 variants are absent from even large metaanalysis studies of VTE risk.5,11,12 Our VTE study did not include rs141041254 because its allele frequency was >0.05%. When the distribution for rs141041254 was looked at, there was not a significant difference between cases vs controls, although we were very underpowered for single-variant tests (OR = 2.09, Fisher's exact 2-sided, P = .4155). The lack of a “genome-wide” significant signal at STAB2 in VTE GWAS supports the idea that large effect size is required, and many other loci with allelic heterogeneity for rare variants may have important roles in complex genetic traits even though large GWAS may be unable to detect these loci.48
Even though STAB2 has not been directly associated with VTE in previous studies, it remains a strong candidate gene for VTE. The link between variants in STAB2 and plasma VWF levels is strengthened by a previous study of murine Stab2 deficiency that demonstrated stabilin-2 functions as a clearance receptor for VWF.38 An earlier study in a large French Canadian family with VTE detected a linkage signal near the STAB2 locus for plasma VWF levels.4 Our study using TwinsUK plasma and DNA sequencing data demonstrates the association between rare variants in STAB2 and plasma VWF levels in humans consistent with common variant associations identified in GWAS. Interestingly, 4 individuals in the TwinsUK study had rare variants we also detected in our VTE cases and characterized in cell culture models (supplemental Table 7).
As a scavenger receptor, we expect stabilin-2 to remove multiple different components from blood. Our study and others have made a connection between stabillin-2 and plasma VWF levels. However, existing literature provides evidence that stabilin-2 is an important clearance receptor for a large number of nonprotein ligands such as hyaluronic acid, heparin sulfate, extracellular DNA, and phosphatidylserine-exposed cell membranes, suggesting that there may be multiple changes in the plasma of an individual with decreased stabilin-2 function that alter VTE risk. Without a comprehensive understanding of these ligands, we are limited in our ability to design a specific stabilin-2 functional assay for VTE risk. We hypothesize that many STAB2 variants operate through altered cell surface expression of stabilin-2 due to protein misfolding or nonsense mediated decay, which would inhibit the interaction of stabilin-2 and any of its ligands. Our analysis of stable cell lines expressing stabilin-2 variants generally supports this hypothesis by demonstrating increased retention of variants in the ER and decreased surface expression compared with reference sequence stabilin-2.
This report describes the role of rare variants in the genetic risk for a common disorder of thrombosis, VTE. The association of STAB2 and VTE was further explored in vitro with a cell culture model. An independent study in the TwinsUK study strengthened the role of stabilin-2 and VWF clearance. Taken together, this report contributes to the comprehensive understanding of the genetic determinants of VTE by the addition of a new candidate gene for VTE risk.
Presented in abstract form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9-12 December 2017 (Best of ASH Award).
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the participants of the ELATE, DODS, GIFT, TwinsUK, and the Institute for Genomic Medicine studies for the use of their samples in this study and Maritjn Van Der Ent for his R coding skills used in Figure 1.
This study was financially supported by the EPIDEMIOM-VT Chair of Excellence from the University of Bordeaux (D.-A.T.). TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, the National Institute for Health Research–funded BioResource, Clinical Research Facility, and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London. This research was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R35 HL135793 (D. Ginsburg) and R01HL141399 (K.C.D.), and the University of Michigan Department of Pediatrics Charles Woodson Accelerator award (K.C.D.). K.C.D. is a member of the Cell and Molecular Biology Program at the University of Michigan. D. Ginsburg is a Howard Hughes Investigator.
Authorship
Contribution: K.C.D., A.B.O., M.H., D.-A.T., P.H.R., C.K., J.B.R., J.Z.L., D. Goldstein, and D. Ginsburg designed the study; P.H.R., C.K., F.W., and D. Goldstein contributed participant samples; K.C.D., A.B.O., P.M.J., K.G., and M.U. performed the bench research; K.C.D., A.B.O., M.H., M.G., D.-A.T., L.M., and J.B.R. analyzed the data; K.C.D., A.B.O., M.H., and D. Ginsburg wrote one or more parts of the manuscript; and all authors reviewed and provided comments on the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Karl C. Desch, University of Michigan, Department of Pediatrics, 1150 W Medical Center Dr, MSRB III Bldg, Room 8315, Ann Arbor, MI 48109; e-mail: kdesch@med.umich.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal