

Key Points
FcγRIIA, an IC receptor, promotes nephritis and thrombosis in lupus.
FcγRIIA expression modifies the platelet transcriptome and accelerates platelet activation in lupus.

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune inflammatory disease characterized by deposits of immune complexes (ICs) in organs and tissues. The expression of FcγRIIA by human platelets, which is their unique receptor for immunoglobulin G antibodies, positions them to ideally respond to circulating ICs. Whereas chronic platelet activation and thrombosis are well-recognized features of human SLE, the exact mechanisms underlying platelet activation in SLE remain unknown. Here, we evaluated the involvement of FcγRIIA in the course of SLE and platelet activation. In patients with SLE, levels of ICs are associated with platelet activation. Because FcγRIIA is absent in mice, and murine platelets do not respond to ICs in any existing mouse model of SLE, we introduced the FcγRIIA (FCGR2A) transgene into the NZB/NZWF1 mouse model of SLE. In mice, FcγRIIA expression by bone marrow cells severely aggravated lupus nephritis and accelerated death. Lupus onset initiated major changes to the platelet transcriptome, both in FcγRIIA-expressing and nonexpressing mice, but enrichment for type I interferon response gene changes was specifically observed in the FcγRIIA mice. Moreover, circulating platelets were degranulated and were found to interact with neutrophils in FcγRIIA-expressing lupus mice. FcγRIIA expression in lupus mice also led to thrombosis in lungs and kidneys. The model recapitulates hallmarks of human SLE and can be used to identify contributions of different cellular lineages in the manifestations of SLE. The study further reveals a role for FcγRIIA in nephritis and in platelet activation in SLE.
Introduction
Systemic lupus erythematosus (SLE) affects ∼1 in 1000 individuals, mostly women.1 Autoimmunity in SLE involves aberrant activation of the immune system in response to circulating autoantigens (eg, nuclear proteins and DNA) and is characterized by increased levels of type I interferon (eg, IFN-α).2-4 Circulating autoantibodies recognize autoantigens and form immune complexes (ICs). IC formation leads to their deposition in tissues, thus promoting the breakdown of immune tolerance and the initiation of cellular activation.5-7 Hence, inflammation affects the connective tissues and blood vessels of many organs and systems, such as the kidneys, lungs, skin, joints, and central nervous system.2 Patients with SLE are also more prone to thrombosis (pulmonary embolism and deep vein thrombosis) and to lethal cardiovascular diseases.8,9 Up to 15% of patients develop persistent thrombocytopenia,10-12 which is generally associated with a poor prognosis.
Platelets are anucleate cells released by megakaryocytes. They patrol the blood circulation to ensure blood vessel integrity13 but are also equipped with a complex network of immune receptors and inflammatory molecules that are packaged into their granules and released upon platelet activation, suggesting an active role for platelets in inflammatory diseases.14,15 In SLE, platelets present surface P-selectin and have a reduced content of serotonin, indicating the release of alpha (α) and dense (δ) granule components by activated platelets.9,14 Extracellular vesicles (EVs), small membrane-bound vesicles that can transport platelet-derived mediators (eg, cytokines, RNA, enzymes, lipid mediators), are produced by activated platelets and are increased in blood of patients with SLE.16 Furthermore, soluble platelet-derived inflammatory mediators are detectable in the blood of patients with SLE,17-22 and their content in S100A8/A923 and interleukin-1β,24 pro-inflammatory molecules, increases in SLE platelets.23,24 Moreover, a type I IFN signature is identifiable in both the platelet transcriptome and proteome during SLE, especially in patients with a history of vascular diseases.25
Immunoglobulin G (IgG)-containing ICs represent the main form of ICs found in SLE.26 Humans express 6 members of the FcγR family (FcγRI, FcγRIIA, FcγRIIB, FcγRIIC, FcγRIIIA, and FcγRIIIB [the latter lacking an intracellular signaling domain]).27 FcγR display different affinities for IgG subclasses and can all transduce activating signals except FcγRIIB, which is considered an inhibitory FcγR.28 Ablation of the common FcRγ chain,29 or murine FcγR,30 protects against SLE in mice, whereas ablation of FcγRIIB exacerbates SLE.31 However, the exact contribution of each individual FcγR to SLE, and whether they play a protective or a deleterious role in SLE, remains unknown.
In particular, FcγRIIA is a low-affinity activatory receptor and its polymorphism was suggested to increase susceptibility to renal manifestations in SLE.32,33 FcγRIIA is expressed by platelets (and megakaryocytes), neutrophils, monocytes, macrophages, mast cells, and dendritic cells in humans.34,35 FcγRIIA is the sole FcγR expressed by platelets in humans and, as a consequence of platelet surplus in the circulation, is the most abundantly expressed FcγR in blood.34 Studies on human platelets show that ICs from patients with SLE can activate platelets through FcγRIIA.17 Moreover, human platelets efficiently endocytose ICs, suggesting that they may contribute to the clearance of ICs and may thereby dampen inflammation in SLE.36,37
FcγRIIA is absent in mice, and thus murine platelets are devoid of any FcγR capable of recognizing IgG and are completely irresponsive to ICs.27,35 This contrasts with murine leukocytes, which do express other members of the FcγR family and can therefore still respond to ICs.35,38,39 Thus, the sequence of events in current murine models of circulating ICs is biased, as it strongly favors leukocytes and dismisses platelets, arguably the most important cell population capable of recognizing ICs in humans. The comprehensive impact of platelets in murine SLE has therefore not yet been fully explored, as previous studies found them to solely play a supporting role in response to later-stage events such as organ damage and complement cascade activation.2,40 Transgenic expression of FcγRIIA (FcγRIIATGN) was introduced in C57BL/6J mice.41 FcγRIIA in platelets in these mice signals through spleen tyrosine kinase42,43 and requires the activities of guanine nucleotide exchange factor CalDAG-GEF144 and 12-lipoxygenase.38 FcγRIIATGN mice were successfully used to study the role of FcγRIIA in acute models of ICs, such as heparin-induced thrombocytopenia,45 anaphylaxis,46 and sepsis.47 These studies showed that platelets play a lead role during acute exposure to ICs when the FcγRIIA transgene is expressed.46,47
By adding the FCGR2A transgene to an SLE lupus model, we were able to examine its role in lupus pathogenesis, thereby providing an outstanding model for the study of cell activation through this receptor in SLE. We used the model to evaluate platelet activation in SLE.
Methods
Guidelines of the Canadian Council on Animal Care were followed in all mouse studies, and the protocol was approved by the Animal Welfare Committee at Laval University (2017-122-2). Human participants were recruited with an approval from the CHU de Québec Ethics Committee (#B14-08-2108). The study was conducted in accordance with the Declaration of Helsinki.
Mice
FcγRIIATGN (C57BL/6J) hemizygous mice, NZW/LacJ and NZB/BINJ, were purchased from The Jackson Laboratory. NZW/LacJ and FcγRIIATGN mice were backcrossed to obtain NZW/LacJ mice expressing FcγRIIA (NZW/LacJ.FcγRIIATGN). F1 progeny were obtained by crossing the NZW/LacJ.FcγRIIATGN and NZB/BINJ strains. Guidelines of the Canadian Council on Animal Care were followed in all mouse studies.
Additional methods are presented in the supplemental Materials (available on the Blood Web site).
Results
Platelet activation in human SLE
In vitro stimulation of platelets from healthy donors with ICs resulted in the change of αIIbβ3 to its activated conformation (αIIbβ3*) and to the release of CD62P+ EVs (Figure 1A-B). The monoclonal antibody anti-FcγRIIA (clone IV.3) completely blunted the activation, suggesting that upon FcγRIIA signaling, platelets are activated and CD62P translocates from α-granules and is thereby expressed on EVs.
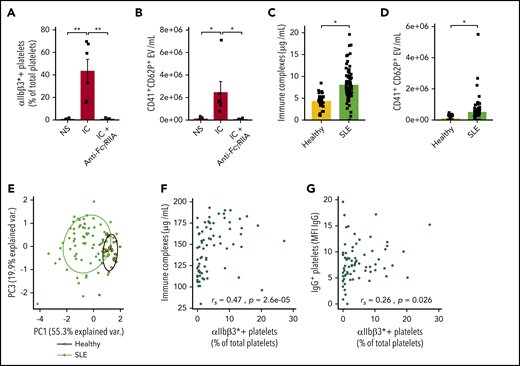
IC-FcγRIIA mediated platelet activation in human SLE. ICs induce platelet activation (A) and CD62P+ microvesicle release (B) in vitro (n = 5). (C) Quantification of serum ICs in patients with SLE (n = 73) and healthy volunteers (n = 30). (D) Quantification of CD41+CD62P+ circulating vesicles in the plasma of patients with SLE (n = 68) and healthy volunteers (n = 30). (E) Principal component (PC) analysis based on 3 variables: platelet activation, IC levels, and IgG+ platelets in patients with SLE and healthy volunteers. Correlation between the levels of activated platelets (αIIbβ3*+) with ICs (F) and with IgG+ platelets (G) in patients with SLE (n = 73). Data are presented as the mean ± SEM. Statistical analyses: 1-way analysis of variance (A-B), Wilcoxon test (C-D), and Spearman’s rank correlation (F-G). *P < .05, **P < .01. NS, nonstimulated.
IC-FcγRIIA mediated platelet activation in human SLE. ICs induce platelet activation (A) and CD62P+ microvesicle release (B) in vitro (n = 5). (C) Quantification of serum ICs in patients with SLE (n = 73) and healthy volunteers (n = 30). (D) Quantification of CD41+CD62P+ circulating vesicles in the plasma of patients with SLE (n = 68) and healthy volunteers (n = 30). (E) Principal component (PC) analysis based on 3 variables: platelet activation, IC levels, and IgG+ platelets in patients with SLE and healthy volunteers. Correlation between the levels of activated platelets (αIIbβ3*+) with ICs (F) and with IgG+ platelets (G) in patients with SLE (n = 73). Data are presented as the mean ± SEM. Statistical analyses: 1-way analysis of variance (A-B), Wilcoxon test (C-D), and Spearman’s rank correlation (F-G). *P < .05, **P < .01. NS, nonstimulated.
The levels of ICs and circulating platelet EVs were then assessed in a cohort of patients with SLE and healthy individuals (supplemental Table 1), and the CD62P marker was used to distinguish platelet-derived EVs from those derived from megakaryocytes.48 IgG-containing ICs were higher in patients with SLE (Figure 1C). The levels of CD62P+ platelet EVs (CD41+CD62P+ EVs) were also increased in the blood circulation of patients with SLE, consistent with the previously reported increase in platelet EVs in SLE49 (Figure 1D). Principal component analysis based on the expression of αIIbβ3* and the presence of plasma ICs and of IgG at the platelet surface showed that although healthy donors formed a homogeneous cluster, patients with SLE clearly segregated from healthy donors and displayed a higher variability, consistent with the heterogeneity of this disease (Figure 1E). Interestingly, the number of αIIbβ3* platelets correlated with the concentrations of circulating ICs and with the presence of IgG on platelets (Figure 1G-H). Thus, the data support a significant role for ICs in platelet activation and the interaction of activated platelets with the prevailing ICs in SLE.
Generation of an SLE model expressing FcγRIIA
Male mice from the previously reported transgenic strain on the C57BL/6 background (C57BL/6.FcγRIIATGN), which expresses human FcγRIIA on myeloid cells (as in humans),41 were backcrossed with NZW/LacJ female mice. Offspring closest to the NZW/LacJ (≥95%) (supplemental Table 2) were selected and backcrossed for further generations to reach 99% NZW/LacJ background. The resulting male NZW/LacJ mice were used for breeding with NZW/BINJ female mice to generate the F1 generation composed of mice expressing FcγRIIA (B/WF1.FcγRIIATGN) or not (B/WF1.FcγRIIANull). Analysis of the F1 confirmed that FcγRIIA was transmitted according to Mendelian segregation.
As in humans and in C57BL/6.FcγRIIATGN mice,41 FcγRIIA was detected in the bone marrow on the surface of myeloid cells (CD11b+), whereas its expression was low/undetectable on B and T lymphocytes (supplemental Figure 1A). Analysis using an antibody cocktail to exclude differentiated lineage progenitors (lymphocytes, monocytes/macrophages, NK cells, erythrocytes, and granulocytes) showed that FcγRIIA was also expressed by a fraction (3.1 ± 1.3%) of Lin– cells. The latter population included megakaryocyte progenitors, which represented 23.5 ± 7% of the total Lin– FcγRIIA+ cells (supplemental Figure 1B). FcγRIIA was found on the surface of all circulating platelets (99.9 ± 0.1%), with a stable intensity (supplemental Figure 1C). Whole mouse immunofluorescence analysis of B/WF1.FcγRIIATGN mice confirmed the presence of FcγRIIA+CD41+ and FcγRIIA+CD11b+ cells in the bone marrow but also in lymph nodes, liver, kidneys, the intestine, and in the meninges surrounding the brain (Figure 2A).

SLE murine model expressing human FcγRIIA. (A) Whole body sagittal-section of a B/WF1.FcγRIIATGN mouse (female, 28 weeks) with SLE showing FcγRIIA staining in CD11b+ cells (white arrows) and in CD41+ megakaryocytes (white arrow heads) in the brain (b), lymph node (c), salivary gland (d), lungs (e), liver (f), kidney (g), intestine (h), and bone marrow (i). Scale bars, 5 mm (A); 50 µm (b-i). The illustrated image is a composite of all scanned tissue areas automatically generated. Areas not presenting tissues were automatically filled in black to generate a clearer image. (B) Levels of total serum IgG in B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN female mice from 10 to 30 weeks. N = 5 to 8 per group and per time point. (C) Plasma B cells in the spleen of B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice at 28 weeks of age (n = 5). Data are presented as the mean ± SEM. Statistical analyses: 2-way analysis of variance. (B) Šídák’s multiple comparisons test for comparing B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN, then Dunnett’s multiple comparisons test to compare each strain to 10 weeks. (C) Mann-Whitney U test (comparing B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN). *P < .05, **P < .01.
SLE murine model expressing human FcγRIIA. (A) Whole body sagittal-section of a B/WF1.FcγRIIATGN mouse (female, 28 weeks) with SLE showing FcγRIIA staining in CD11b+ cells (white arrows) and in CD41+ megakaryocytes (white arrow heads) in the brain (b), lymph node (c), salivary gland (d), lungs (e), liver (f), kidney (g), intestine (h), and bone marrow (i). Scale bars, 5 mm (A); 50 µm (b-i). The illustrated image is a composite of all scanned tissue areas automatically generated. Areas not presenting tissues were automatically filled in black to generate a clearer image. (B) Levels of total serum IgG in B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN female mice from 10 to 30 weeks. N = 5 to 8 per group and per time point. (C) Plasma B cells in the spleen of B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice at 28 weeks of age (n = 5). Data are presented as the mean ± SEM. Statistical analyses: 2-way analysis of variance. (B) Šídák’s multiple comparisons test for comparing B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN, then Dunnett’s multiple comparisons test to compare each strain to 10 weeks. (C) Mann-Whitney U test (comparing B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN). *P < .05, **P < .01.
FcγRIIA is dispensable in autoantibody production and immune cell proliferation
B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice developed comparable levels of total circulating IgG (Figure 2B) and similar levels of splenic plasma B (CD19+ CD138+) cells at 28 weeks (Figure 2C), by which time both strains displayed the highest level of serum IgG. Moreover, both strains equally developed IgG autoantibodies that targeted double-stranded DNA and nuclear proteins Ro/SSA (anti–Ro/SSA) and La/SSB (anti–La/SSB), except those targeting ribonuclear proteins (anti–Sm/RNP) and anti-Smith (anti-Sm) that were higher in B/WF1.FcγRIIATGN mice at 28 weeks (supplemental Figure 2A-E).
The proportions of bone marrow T and B lymphocytes and neutrophils were also comparable (supplemental Figure 2F), suggesting that FcγRIIA is not involved in the proliferation of a particular immune cell population.
FcγRIIA accelerates SLE
Despite the comparable levels of autoantibodies, B/WF1.FcγRIIATGN mice exhibited impaired survival, and 50% of B/WF1.FcγRIIATGN female mice died at 28 weeks, compared with 38 weeks for B/WF1.FcγRIIANull mice (Figure 3A). Similar results were observed in male mice (Figure 3B), although the difference between the 2 strains was less striking (50% survival, 41 weeks in B/WF1.FcγRIIATGN and 46 weeks in B/WF1.FcγRIIANull littermates). In both female and male mice, death occurred in mice showing severe urine protein (proteinuria), a marker of kidney failure (supplemental Figure 3A). Thus, FcγRIIA accelerates SLE-related morbidity but preserves the sex bias toward females as in human SLE and the B/WF1 model.2 Consistent with these observations, further analyses were restricted to female mice.
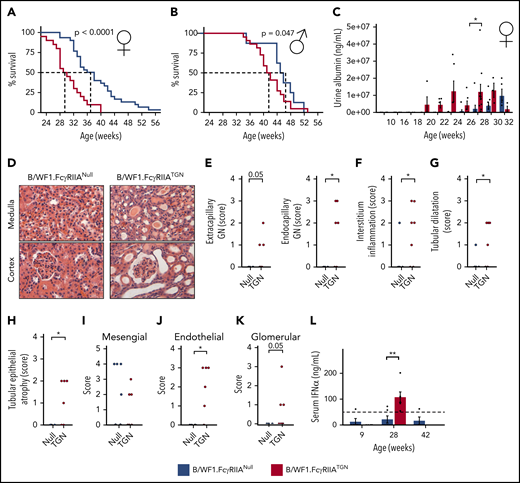
FcγRIIA accelerates SLE development in the B/WF1.FcγRIIATGNmouse model. Survival analysis of female (n = 49) (A) and male (n = 33) (B) B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice. (C) Evolution of urine albumin in female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice aged 10 to 32 weeks; n = 5 to 8 per group. (D) Kidney histology, hematoxylin and eosin staining of female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN kidneys at 28 weeks. Representative images of n = 5 per group. Original magnification ×100. Histological assessment of: extracapillary and endocapillary glomerulonephritis (GN) (E), interstitial inflammation (F), kidney tubular dilatation (G), and epithelial atrophy (H) in female B/WF1.FcγRIIATGN mice (n = 6) and their age-matched B/WF1.FcγRIIANull littermates (n = 5). (I-K) Analysis of IgG distribution in the kidneys according to electron microscopy. (L) Quantification of serum IFN-α in female B/WF1.FcγRIIATGN mice and their age-matched B/WF1.FcγRIIANull littermates (n = 4-5). Data are presented as the mean ± SEM (C,L), or mean with median (E-I). Statistical analyses: log-rank survival analysis (A-B), 2-way analysis of variance, Šídák’s multiple comparisons test (C), unpaired Student t test (E-K), and Mann-Whitney U test (L). *P < .05, **P < .01. P = .05 are indicated.
FcγRIIA accelerates SLE development in the B/WF1.FcγRIIATGNmouse model. Survival analysis of female (n = 49) (A) and male (n = 33) (B) B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice. (C) Evolution of urine albumin in female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice aged 10 to 32 weeks; n = 5 to 8 per group. (D) Kidney histology, hematoxylin and eosin staining of female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN kidneys at 28 weeks. Representative images of n = 5 per group. Original magnification ×100. Histological assessment of: extracapillary and endocapillary glomerulonephritis (GN) (E), interstitial inflammation (F), kidney tubular dilatation (G), and epithelial atrophy (H) in female B/WF1.FcγRIIATGN mice (n = 6) and their age-matched B/WF1.FcγRIIANull littermates (n = 5). (I-K) Analysis of IgG distribution in the kidneys according to electron microscopy. (L) Quantification of serum IFN-α in female B/WF1.FcγRIIATGN mice and their age-matched B/WF1.FcγRIIANull littermates (n = 4-5). Data are presented as the mean ± SEM (C,L), or mean with median (E-I). Statistical analyses: log-rank survival analysis (A-B), 2-way analysis of variance, Šídák’s multiple comparisons test (C), unpaired Student t test (E-K), and Mann-Whitney U test (L). *P < .05, **P < .01. P = .05 are indicated.
Analysis of proteinuria over time revealed that alterations in kidney function appeared at 20 weeks in B/WF1.FcγRIIATGN mice, ∼8 weeks earlier than in their B/WF1.FcγRIIANull littermates (Figure 3C). It is noteworthy that the apparent fluctuations in proteinuria in B/WF1.FcγRIIATGN mice (eg, weeks 26 and 32) are due to the death of mice with the most severe kidney damage.
Proteinuria concurred with impaired kidney histology at 28 weeks (Figure 3D). Extracapillary glomerulonephritis (cell proliferation and immune infiltrate in Bowman’s space),50 endocapillary glomerulonephritis (cell proliferation and immune infiltrate in the glomerular basement membrane) (Figure 3E), and signs of interstitial nephritis (Figure 3F) were only present in B/WF1.FcγRIIATGN mice at 28 weeks. These mice also displayed other lesions characteristic of nephritis, such as tubular dilatation and epithelial atrophy (Figure 3G-H), which were absent in age-matched B/WF1.FcγRIIANull mice. An IgG infiltrate was observed in the kidneys of B/WF1.FcγRIIATGN and B/WF1.FcγRIIANull mice. However, although IgG was equally present in the kidney mesangial zone of both strains (Figure 3I), it was only visible in the endothelial and glomerular zones in B/WF1.FcγRIIATGN mice (Figure 3J-K), suggesting that the disease progressed to end-stage nephritis in the presence of FcγRIIA. It is worth noting that B/WF1.FcγRIIANull mice aged between 35 and 45 weeks exhibited impaired kidney histology and kidney IgG similar to that of B/WF1.FcγRIIATGN mice (supplemental Figure 3B), which suggests that FcγRIIA in fact accelerated nephritis manifestations. Circulating IFN-α, a pro-inflammatory cytokine highly pathogenic in human SLE,2 was detected in the plasma of B/WF1.FcγRIIATGN mice but was undetectable in B/WF1.FcγRIIANull mice, even when they later developed SLE (Figure 3L). This scenario is consistent with its reported absence in chronic murine models of SLE.51
Role of bone marrow in FcγRIIA-mediated acceleration of SLE
Given the striking promotion of nephritis by FcγRIIA expression, kidneys were investigated. Immuno-histochemistry revealed that although FcγRIIA expression was nearly absent in healthy kidneys from B/WF1.FcγRIIATGN mice (9 weeks), FcγRIIA was detectable in kidneys, notably in the glomeruli and tubules of mice aged 28 to 29 weeks and displaying SLE symptoms (Figure 4A).
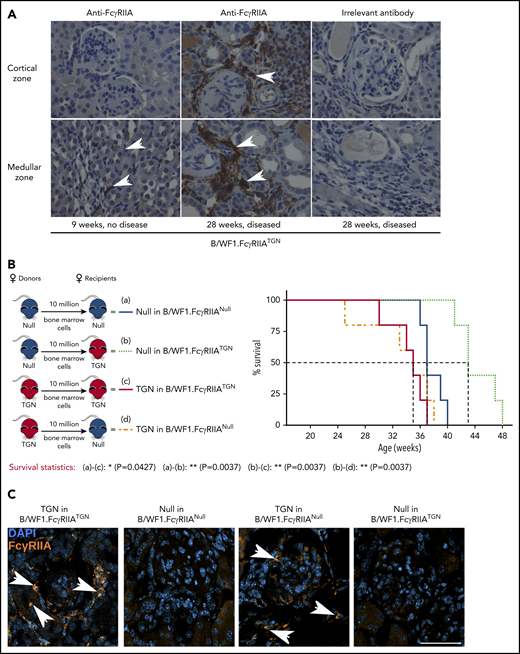
Pathogenic FcγRIIA is provided by the bone marrow. (A) Immunohistochemical staining of FcγRIIA protein (white arrows) in the kidneys of female B/WF1.FcγRIIATGN mice aged 9 and 28 weeks (×400 magnification). Data are representative of n = 5 per group. (B) Left panel: schematic of bone marrow chimera expressing or not FcγRIIA. Right panel: survival analysis of bone marrow chimera expressing or not FcγRIIA on bone marrow cells (n = 5 per group). (C) Kidney immunofluorescence staining in bone marrow chimera with SLE. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue), and white arrows indicate FcγRIIA+ cells (orange). Single staining is presented in supplemental Figure 5. Scale bar, 50 µm. (B) Statistical analyses, log-rank survival analysis. *P < .05, **P < .01.
Pathogenic FcγRIIA is provided by the bone marrow. (A) Immunohistochemical staining of FcγRIIA protein (white arrows) in the kidneys of female B/WF1.FcγRIIATGN mice aged 9 and 28 weeks (×400 magnification). Data are representative of n = 5 per group. (B) Left panel: schematic of bone marrow chimera expressing or not FcγRIIA. Right panel: survival analysis of bone marrow chimera expressing or not FcγRIIA on bone marrow cells (n = 5 per group). (C) Kidney immunofluorescence staining in bone marrow chimera with SLE. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue), and white arrows indicate FcγRIIA+ cells (orange). Single staining is presented in supplemental Figure 5. Scale bar, 50 µm. (B) Statistical analyses, log-rank survival analysis. *P < .05, **P < .01.
Because FcγRIIA is expressed by the myeloid lineage, we examined the contribution of bone marrow–derived lineages to lupus nephritis using bone marrow chimera. B/WF1.FcγRIIANull recipients engrafted with B/WF1.FcγRIIATGN cells (TGN in B/WF1.FcγRIIANull) exhibited the same 50% survival (34 weeks) as control B/WF1.FcγRIIATGN mice engrafted with B/WF1.FcγRIIATGN cells (TGN in B/WF1.FcγRIIATGN) (Figure 4B). Conversely, the absence of FcγRIIA in bone marrow–derived cells in B/WF1.FcγRIIATGN mice engrafted with bone marrow from B/WF1.FcγRIIANull mice (TGN in B/WF1.FcγRIIANull) led to prolonged survival. An intriguing observation is that the transfer of bone marrow from B/WF1.FcγRIIANull mice into irradiated B/WF1.FcγRIIATGN mice showed that the expression of FcγRIIA by a radioresistant cell or the mesenchyme might protect mice from lupus.
Analysis of kidneys in these mice revealed the presence of FcγRIIA+ cells when FcγRIIA was expressed by the bone marrow (Figure 4C). Moreover, FcγRIIA was absent in kidneys from B/WF1.FcγRIIATGN mice engrafted with bone marrow from B/WF1.FcγRIIANull mice (TGN in B/WF1.FcγRIIANull), suggesting that FcγRIIA protein identified in kidneys is principally derived from cells originating from the bone marrow and suffice to amplify inflammation.
Upregulation of platelet transcripts involved in a type I IFN response by FcγRIIA
Because FcγRIIA is the exclusive receptor for ICs on the surface of human platelets, with no functional equivalent in mice,34 the addition of FcγRIIA to the B/WF1.FcγRIIA mouse model made it possible to study chronic platelet response in SLE.
Platelet RNA content in all strains was compared at 2 time points: at a predisease state (9 weeks) and at 28 weeks, when ∼50% of the BWF1.FcγRIIATGN mice died of SLE-related complications. By this age, both B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice exhibited a significantly altered platelet transcriptome compared with that of 9-week-old mice (Figure 5A). Because the platelet transcriptome often mirrors that of megakaryocytes,52,53 this finding suggests that megakaryocyte regulatory pathways are responsive to the onset of disease but that they can also occur independently of FcγRIIA expression. However, there were more differentially expressed transcripts identified in B/WF1.FcγRIIATGN mice at 28 vs 9 weeks (377 genes increased and 389 decreased), compared with B/WF1.FcγRIIANull mice at 28 vs 9 weeks (154 increased and 97 decreased).
![FcγRIIA modulates the platelet transcriptome during SLE. (A) Volcano plots of RNA sequencing–based analyses of the platelet transcriptome from B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice aged 9 and 28 weeks (SLE develops by 28 weeks). Red dots represent significantly upregulated (false discovery rate [FDR] <0.05) genes, and blue dots represent significantly downregulated (FDR <0.05) genes. (B) Fold changes for genes contributing to an IFN-α/IFN-β pathway enrichment (FDR = 0.01) in B/WF1.FcγRIIATGN mice aged 28 vs 9 weeks compared with respective fold changes in B/WF1.FcγRIIANull mice aged 28 vs 9 weeks. Gene label H2- refers to the mean fold change for 9 different MHC loci that increased: H2-Bl, D1, K1, Q1, Q10, Q2, Q4, Q6, and Q7. (C) Gene set enrichment analysis directly comparing platelets from 28-week old B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice identified significant (FDR <0.05) positive enrichment (TGN > Null) for 17 pathways. Shown is the enrichment score (ES) plot for the IFN-α/IFN-β Reactome signaling pathway, which had the largest ES and lowest FDR q value. padj, P value adjusted.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/25/10.1182_blood.2020004974/1/m_bloodbld2020004974f5.png?Expires=1763535085&Signature=pLGTL7-yOHsNCaRGLuizdiLHI2FOn4yWw8F9gB1uGJa1Qev74kr6-6pFYVFofkbxR8CR2J0zqXyzayyzBZx-qbxyndh0QtSJeYnOYHi3Y~yMBCotYj24ptGJU86cpuc5pKa4G3kZkZDlbGg0~C8ZLVpZS6IKHu~L2oNLZ1-KknvG6090Yavjl04L1tJXrxWWiF2UqW6kWRyg6j7hGYSvTVdhh5Rg-Wq6aMcmGzoRQi4vfaJznlsgAE~MaAesBQMViKTRYk9u8Fa7636AJHwNvNVzzHEbPFi0sDqD7roq6BY0bm14BtB1X2wbhmHj~daWhYuyqy4rwQ1i1R8qHZ4Oyw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
FcγRIIA modulates the platelet transcriptome during SLE. (A) Volcano plots of RNA sequencing–based analyses of the platelet transcriptome from B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice aged 9 and 28 weeks (SLE develops by 28 weeks). Red dots represent significantly upregulated (false discovery rate [FDR] <0.05) genes, and blue dots represent significantly downregulated (FDR <0.05) genes. (B) Fold changes for genes contributing to an IFN-α/IFN-β pathway enrichment (FDR = 0.01) in B/WF1.FcγRIIATGN mice aged 28 vs 9 weeks compared with respective fold changes in B/WF1.FcγRIIANull mice aged 28 vs 9 weeks. Gene label H2- refers to the mean fold change for 9 different MHC loci that increased: H2-Bl, D1, K1, Q1, Q10, Q2, Q4, Q6, and Q7. (C) Gene set enrichment analysis directly comparing platelets from 28-week old B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice identified significant (FDR <0.05) positive enrichment (TGN > Null) for 17 pathways. Shown is the enrichment score (ES) plot for the IFN-α/IFN-β Reactome signaling pathway, which had the largest ES and lowest FDR q value. padj, P value adjusted.
FcγRIIA modulates the platelet transcriptome during SLE. (A) Volcano plots of RNA sequencing–based analyses of the platelet transcriptome from B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice aged 9 and 28 weeks (SLE develops by 28 weeks). Red dots represent significantly upregulated (false discovery rate [FDR] <0.05) genes, and blue dots represent significantly downregulated (FDR <0.05) genes. (B) Fold changes for genes contributing to an IFN-α/IFN-β pathway enrichment (FDR = 0.01) in B/WF1.FcγRIIATGN mice aged 28 vs 9 weeks compared with respective fold changes in B/WF1.FcγRIIANull mice aged 28 vs 9 weeks. Gene label H2- refers to the mean fold change for 9 different MHC loci that increased: H2-Bl, D1, K1, Q1, Q10, Q2, Q4, Q6, and Q7. (C) Gene set enrichment analysis directly comparing platelets from 28-week old B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice identified significant (FDR <0.05) positive enrichment (TGN > Null) for 17 pathways. Shown is the enrichment score (ES) plot for the IFN-α/IFN-β Reactome signaling pathway, which had the largest ES and lowest FDR q value. padj, P value adjusted.
Although type I IFN genes increased in both B/WF1.FcγRIIATGN and B/WF1.FcγRIIANull mice, the presence of FcγRIIA amplified the IFN response (Figure 5B). Directly comparing 28-week-old B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice confirmed a differential increase in type I IFN pathway genes in transgenic mice (Figure 5C), suggesting that FcγRIIA expression results in alterations in the platelet transcriptome during SLE.
FcγRIIA amplifies platelet activation and thrombosis in SLE
Systemic administration of ICs to FcγRIIATGN mice initiates thrombocytopenia,46,47,54 a frequent and misunderstood manifestation of SLE in humans.10,12 B/WF1.FcγRIIATGN and B/WF1.FcγRIIANull mice had similar platelet counts, which contrasts with acute models of exposure to ICs47,54-56 (Figure 6A).
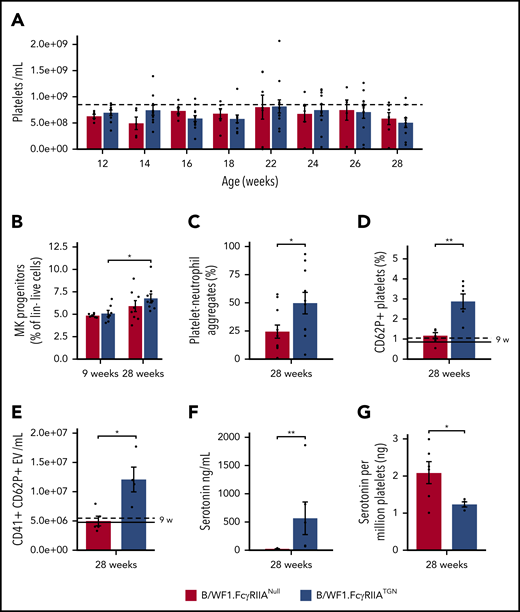
FcγRIIA amplifies chronic platelet activation in SLE. (A) Platelet count in B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN female mice between age 12 and 28 weeks (n = 5 to 8 per group). Dashed line indicates platelet count in 42-week-old C57BL/6.FcγRIIATGN control mice. (B) Analysis of the proportions of megakaryocyte progenitors (MK; selected as lineage-negative, DNA-positive cells expressing CD41 and CD9) in the bone marrow of female B/WF1.FcγRIIA mice at age 9 and 28 weeks (n = 7-9 per group). (C) Quantification of platelet-neutrophil aggregates in the blood of female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice, (n = 11 Null and 10 TGN). Quantification of platelet activation (CD62P+) (D) and CD41+CD62P+ EV (E) in the blood of female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice aged 28 weeks (n = 6 per group). Dashed (B/WF1.FcγRIIATGN) and solid (B/WF1.FcγRIIANull) lines indicate mean of values at 9 weeks (9 w). (F) Quantification of plasma serotonin in female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice at 28 weeks (n = 6). (G) Quantification of platelet serotonin content in female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice with SLE (n = 4-5 per group). Data are presented as the mean ± SEM. Statistical analyses, 2-way analysis of variance with Šídák’s multiple comparisons test (A-B), Mann-Whitney U test (C,D,F), and Student t test (E,G). *P < .05, **P < .01.
FcγRIIA amplifies chronic platelet activation in SLE. (A) Platelet count in B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN female mice between age 12 and 28 weeks (n = 5 to 8 per group). Dashed line indicates platelet count in 42-week-old C57BL/6.FcγRIIATGN control mice. (B) Analysis of the proportions of megakaryocyte progenitors (MK; selected as lineage-negative, DNA-positive cells expressing CD41 and CD9) in the bone marrow of female B/WF1.FcγRIIA mice at age 9 and 28 weeks (n = 7-9 per group). (C) Quantification of platelet-neutrophil aggregates in the blood of female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice, (n = 11 Null and 10 TGN). Quantification of platelet activation (CD62P+) (D) and CD41+CD62P+ EV (E) in the blood of female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice aged 28 weeks (n = 6 per group). Dashed (B/WF1.FcγRIIATGN) and solid (B/WF1.FcγRIIANull) lines indicate mean of values at 9 weeks (9 w). (F) Quantification of plasma serotonin in female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice at 28 weeks (n = 6). (G) Quantification of platelet serotonin content in female B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice with SLE (n = 4-5 per group). Data are presented as the mean ± SEM. Statistical analyses, 2-way analysis of variance with Šídák’s multiple comparisons test (A-B), Mann-Whitney U test (C,D,F), and Student t test (E,G). *P < .05, **P < .01.
B/WF1.FcγRIIATGN mice had more numerous megakaryocyte progenitors (CD41+ Lin–) in the bone marrow at 28 weeks than 9 weeks (Figure 6B), but no differences were observed between B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice. Splenomegaly was also equally observed in both B/WF1.FcγRIIATGN and B/WF1.FcγRIIANull mice aged 28 weeks, although a significant increase in spleen weight was observed between 9 and 28 weeks only in B/WF1.FcγRIIATGN mice (supplemental Figure 4).
Circulating degranulated platelets and platelet-leukocyte aggregates have been reported in patients with SLE.57 Transgenic expression of FcγRIIA led to the formation of platelet-neutrophil aggregates (Figure 6C). Moreover, platelets from B/WF1.FcγRIIATGN mice (28 weeks) revealed an increase in the levels of surface CD62P, indicating translocation of this molecule from α-granule to the cell surface (Figure 6D). Plasma levels of CD41+CD62P+ EVs (Figure 6E) and serotonin (Figure 6F) were also increased, indicating platelet degranulation in the circulation. Because blood serotonin is mainly stored in platelet-dense granules,44 the platelet serotonin content was investigated. B/WF1.FcγRIIATGN platelets had a reduced content in serotonin compared with platelets from diseased B/WF1.FcγRIIANull mice (Figure 6G).
Thrombosis is more frequent in patients with SLE and is an obvious manifestation in the lungs of FcγRIIATGN mice from C57BL/6 background when injected with ICs.46,47 Although thrombosis was not detected in younger mice (Figure 7), histology sections showed a significant increase in thrombi, visible in the microvasculature of the lungs and kidneys in B/WF1.FcγRIIATGN mice when they developed lupus (28 weeks) but not in their age-matched B/WF1.FcγRIIANull littermates. The FcγRIIA expression in fact accelerated thrombosis, as thrombi were observed in B/WF1.FcyRIIANull mice at a later stage, when they too presented kidney nephritis.

The role of FcγRIIA in organ thrombosis in SLE. Representative images of hematoxylin and eosin staining for lung (A) and kidney (B) sections from B/WF1.FcγRIIATGN or B/WF1.FcγRIIANull mice at 9 and 28 weeks of age. Higher magnifications are presented in insets for each condition (scale bars are identified in upper left microscopic images in A and B and are equivalent to 50 µm). Quantification of lung thrombosis (n = 5 per group) (A) and kidney thrombosis (n = 5 per group) (B) in B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice. Arrow heads indicate vascular thrombi, and stars indicate tubule obstruction. Data are presented as the mean ± SEM. Statistical analyses: Šídák’s multiple comparisons test (A-B). Note that in both A and B, a significant difference between 9 and 28 weeks was observed in thrombi counts only in B/WF1.FcγRIIATGN mice. **P < .01, ***P < .001.
The role of FcγRIIA in organ thrombosis in SLE. Representative images of hematoxylin and eosin staining for lung (A) and kidney (B) sections from B/WF1.FcγRIIATGN or B/WF1.FcγRIIANull mice at 9 and 28 weeks of age. Higher magnifications are presented in insets for each condition (scale bars are identified in upper left microscopic images in A and B and are equivalent to 50 µm). Quantification of lung thrombosis (n = 5 per group) (A) and kidney thrombosis (n = 5 per group) (B) in B/WF1.FcγRIIANull and B/WF1.FcγRIIATGN mice. Arrow heads indicate vascular thrombi, and stars indicate tubule obstruction. Data are presented as the mean ± SEM. Statistical analyses: Šídák’s multiple comparisons test (A-B). Note that in both A and B, a significant difference between 9 and 28 weeks was observed in thrombi counts only in B/WF1.FcγRIIATGN mice. **P < .01, ***P < .001.
Discussion
In the current study, we provide a new model of SLE that considers FcγRIIA, and we reveal new insights into the role of FcγRIIA in chronic SLE pathogenesis as well as changes in platelet gene expression and platelet activation in SLE. The findings suggest that the FcγRIIA blockade, by Fab antibodies for instance, might improve SLE.
The B/WF1 model is one of the oldest spontaneous models of SLE.58,59 It develops an active proliferative glomerulonephritis mediated by ICs,58 in part due to defects in IC clearance and the production of autoantibodies targeting double-stranded DNA and nuclear proteins, reminiscent of those found in human SLE.2 In humans, FcγRIIA is expressed on myeloid cells and platelets and is absent in lymphocytes.35 Although our B/WF1 mouse model expressing FcγRIIA recapitulates the human expression pattern well, FcγRIIA was also detected in a fraction of bone marrow progenitors, possibly due to its expression in early hematopoietic progenitors. As for the FcγRIIA expression in kidneys during nephritis, we confirmed that its source was a cellular lineage(s) from the bone marrow that could have invaded this organ. Both neutrophils and platelets shed FcγRIIA in the presence of ICs.60,61 Although the platelet surface expression of FcγRIIA was maintained throughout the duration of the experiment, it was reduced on platelets in mice with severe nephritis that required euthanasia (supplemental Figure 6). We thus suggest that circulating soluble FcγRIIA might accumulate, together with ICs, in the kidney.
Although complement as well as coagulation factors were not examined in the current study, their interplay may participate in thrombo-inflammation in SLE.62 In the B/WF1 model, ablation of FcγR signaling29 confers complete resistance to SLE despite the accumulation of circulating ICs, pointing to a dominant role of FcγR in the inflammatory process in SLE or to a role for complement downstream FcγRs.29,63
Our analysis reveals that in vivo expression of FcγRIIA accelerates SLE and nephritis. Because the concentration of autoantibodies was maintained when FcγRIIA was expressed, this further suggests that expression in blood of the most abundant FcγR35 is not sufficient to promote IC clearance in vivo. Nephritis was the most obvious disease manifestation accelerated in our model as mice died prematurely when FcγRIIA was expressed and developed severe kidney damage. However, it cannot be excluded that FcγRIIA also modifies other manifestations that do not immediately translate into mortality. Although FcγRIIA expression did not affect the occurrence of arthritis in B/WF1 mice (supplemental Figure 7), FcγRIIA promoted thrombosis in the lungs and kidneys, which suggests that FcγRIIA may contribute to elevated risks of thromboembolism. This model may have utility for future investigations into whether FcγRIIA expression in lupus also affects other systems such as the skin, vasculature, and brain.
FcγRIIA accelerated the deposition of ICs in the kidneys, suggesting that cells expressing FcγRIIA might facilitate transport and/or retention of ICs in kidneys. Another hypothesis is that tissue damage caused by FcγRIIA-expressing cells underlies the exposure of neoantigens and thus facilitates local formation of ICs in kidneys. All cells that express FcyRIIA are candidates to amplify nephritis. For instance, the transgenic expression of FcγRIIA in mice revealed that neutrophils as well as monocytes release platelet-activating factor and play a role in anaphylaxis induced by circulating ICs.64 Moreover, when neutrophils express FcγRIIA as the sole FcγR in a transgenic mouse (FcγRIIATGN/γ−/−), they still invade kidneys when mice are injected with antibodies directed against the glomerular basement membrane, showing that FcγRIIA expression suffices to recruit neutrophils in this organ.65 Dendritic cells may be prone to generate IFN-α if stimulated through FcγRIIA,66 which may also explain the presence of IFN-α detected in blood. In the future, this model can thus be used for the examination of contributions of different cellular lineages in SLE.
Consistent with the role of FcγRIIA in amplification of αIIbβ3 outside-in signaling, activation of αIIbβ3 correlated with concentrations of ICs in SLE patients.42,67 FcγRIIA expression in platelets may thus directly activate platelets following the accumulation of ICs, and can amplify platelet activation due to organ and vessel damage. Platelets may also contribute to inflammation in the absence of FcγRIIA, but at a later stage when injury is evident, which may explain how the depletion of platelets improved nephritis in B/WF1.FcγRIIANull mice.17
Intriguingly, thrombosis was present in the lungs and kidneys, with no perceptible impact on platelet levels in blood. Although further studies are necessary to confirm the presence of platelets in thrombi, perhaps platelets gradually accumulate in the lungs and kidneys in this chronic disease, and mechanisms, such as emergency hematopoiesis in spleen, compensate for the loss of platelets in thrombi. This would be consistent with the splenomegaly (supplemental Figure 4) we observed in lupus mice. Because FcγRIIA did not increase thrombocytopenia, this suggests that other mechanisms may lead to reduced platelet number in certain SLE patients; these mechanisms include immune thrombocytopenia due to platelet-specific autoantibodies, T cell–mediated platelet depletion, or accelerated deglycosylation processes.68,69
The platelet transcriptome was affected by FcγRIIA expression, suggesting that megakaryocytes may respond to ICs (in bone marrow if accessed by ICs, or in the lungs where megakaryocytes are also described).70 Notably, the top enriched pathway in platelets when FcγRIIA was expressed was a type I IFN response. This is consistent with the reported changes to the human platelet transcriptome in SLE,25 which exhibits enrichment in type I IFN response. Intriguingly, the comparison of the transcriptome of the human and murine platelets in SLE revealed significant similarities. A significant overlap was found between genes previously reported to increase in patients with SLE compared with genes changed in 28- vs 9-week-old B/WF1.FcγRIIATGN mice (P = 1.4e-6; hypergeometric test) but not B/WF1.FcγRIIANull mice (P = .49). Genes that overlapped between humans and transgenic mice were enriched in the type I IFN signaling pathway (P = 1.3e-4) (supplemental Figure 8), further highlighting the relevance of the addition of the transgene to recapitulate human features.
During viral infection, megakaryocytes produce IFN-α, which activates bystander megakaryocytes as well as stem cells, thereby promoting transcription of IFN-stimulated genes and promoting antiviral immune responses.71 IFN-α detected in FcγRIIA-expressing lupus mice may therefore affect megakaryocytes, which would explain the upregulation of IFN-related gene expression in platelets that we observed in these mice. Although IFN can be produced by megakaryocytes upon stimulation, it is also possible that CD40L, liberated by IC-stimulated platelets, triggers IFN-α production by dendritic cells.17 Although IFN-α was found uniquely in lupus mice expressing FcγRIIA, not all inflammatory cytokines were increased due to the presence of the transgene. With the exception of tumor necrosis factor and interleukin-15 (of the 32 cytokines evaluated), which were more elevated in mice lacking FcγRIIA, the plasma content in cytokines was similar in both groups of diseased mice (supplemental Figure 9). Whether more obvious variations in cytokine production would be observed in diseased organs was not determined, but the data suggest that although FcγRIIA affects the IFN signature hallmark in SLE, it has only a modest contribution to the overall production of cytokines in blood.
The interactions of platelet with leukocytes and the release of EVs are hallmarks of inflammation and occur in SLE.9 Platelets and neutrophils interact through CD62P or glycoprotein Ib, found on platelets, as well as P-selectin glycoprotein ligand-1 and Mac-1, found on neutrophils.72,73 Fibrinogen also contributes to platelet–neutrophil interactions by bridging αIIbβ3 and Mac-1.74 Activated αIIbβ3 can also bind the solute carrier family 44 member 2 on neutrophils.75 The interactions between platelets and neutrophils observed in FcγRIIA-expressing lupus mice may favor neutrophil migration into tissues. Although αIIbβ3 is activated and CD62P is expressed on platelets in SLE, it remains to be established whether these molecules are implicated in those interactions. Moreover, we revealed the contribution of FcγRIIA in the release of EV in lupus. Because EVs are involved in intercellular communication, the model can be used to identify the EV content and how EVs may contribute to the pathogenesis.
The blockade of CD40L was effective in the SLE mouse model,76 but its use in humans was halted as it triggered thromboembolism in patients with SLE.77 Moreover, therapies targeting kinase pathways (eg, JAK1/2 inhibitors) led to platelet activation in lungs in patients with rheumatoid arthritis, thus suggesting that improved mouse models that permit the evaluation of potential platelet activation in rheumatic diseases are needed. Here, we provide a new murine model of SLE that recapitulates several features of the disease observed in humans. This model will offer a better understanding of the involvement of chronic platelet activation and provides a valuable tool for testing new therapeutic molecules in SLE.
RNA-sequencing data are submitted to NCBI short read archive (SRA) BioProject PRJNA655552.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to the generous technical help provided by Emmanuelle Rollet-Labelle, Charles Joly Beauparlant, and Antoine Bodein throughout the study. The authors thank the Bioimagerie du Petit Animal, the Cytometry, and the Microscopy platforms (CHU de Québec). The authors also thank the High-Throughput Genomics and Bioinformatic Analysis Core at the Huntsman Cancer Institute, University of Utah.
This work was supported by a foundation grant from the Canadian Institutes of Health Research (CIHR) (E.B.). E.B. is recipient of a new investigator award from the CIHR and the Fonds de Recherche en Santé du Québec (FRQS); and is a Canadian National Transplant Research Program researcher. P.R.F. is a recipient of a tier 1 Canada Research Chair on Systemic Autoimmune Rheumatic Diseases. N.T. and I.M. are recipients of fellowships from The Arthritis Society and from FRQS. R.A.C. is supported by a grant from the National Institutes of Health (NIH), National Institute on Aging (K01AG059892). M.T.R. is supported by grants from the NIH, National Heart, Lung, and Blood Institute (HL142804 and HL130541) and the National Institute on Aging (AG048022 and AG059877). M.T.R. was also supported, in part, by Merit Review Award Number I01 CX001696 from the US Department of Veterans Affairs Clinical Sciences R&D Service. This material is, in part, the result of work supported with resources and the use of facilities at the George E. Wahlen Veterans Affairs Medical Center. K.R.M. supported by the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (K01DK111515), and is an American Society of Hematology Scholar. J.R. is a recipient of an operating grant from the CIHR (PJT-159652). D.S. was the recipient of a studentship from FRQS and Merit Fellowships from the Department of Microbiology and Immunology. C.L. is supported by the Lupus Research Alliance (519414).
Authorship
Contribution: I.M., E.B., I.A., N.T., and N.C. conceived and designed experiments; N.P., C.L., J.W.R., M.T.R., A.D., K.R.M., S.L., S.E.M., R.A.C., J.R., and P.R.F. contributed critical reagent, resources, and expertise; I.M., N.T., I.A., B.M., D.S., C.L., T.L., J.W.R., N.C., A.D., A.Z., A.L., and J.R. performed experiments; I.M., N.T., I.A., B.M., N.C., T.L., J.W.R., N.C., A.D., A.Z., A.L., J.R., and N.P. processed and analyzed data; I.M. and E.B. supervised the data processing and analysis; I.M., N.T., and E.B. wrote the manuscript; N.T. provided the visual abstract; and all authors critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Eric Boilard, Centre de Recherche du Centre Hospitalier Universitaire de Québec, Faculté de Médecine de l’Université Laval, 2705 Laurier Blvd, Room T1-49, Québec, QC G1V 4G2, Canada; e-mail: eric.boilard@crchudequebec.ulaval.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal