

Key Points
BM regeneration requires donor hematopoietic progenitor mitochondria transfer to the host mesenchymal microenvironment.
Mitochondrial transfer from donor HSPC to host BM MSC is regulated positively by hematopoietic Cx43 and negatively by hematopoietic AMPK.

Abstract
The fate of hematopoietic stem and progenitor cells (HSPC) is tightly regulated by their bone marrow (BM) microenvironment (ME). BM transplantation (BMT) frequently requires irradiation preconditioning to ablate endogenous hematopoietic cells. Whether the stromal ME is damaged and how it recovers after irradiation is unknown. We report that BM mesenchymal stromal cells (MSC) undergo massive damage to their mitochondrial function after irradiation. Donor healthy HSPC transfer functional mitochondria to the stromal ME, thus improving mitochondria activity in recipient MSC. Mitochondrial transfer to MSC is cell-contact dependent and mediated by HSPC connexin-43 (Cx43). Hematopoietic Cx43-deficient chimeric mice show reduced mitochondria transfer, which was rescued upon re-expression of Cx43 in HSPC or culture with isolated mitochondria from Cx43 deficient HSPCs. Increased intracellular adenosine triphosphate levels activate the purinergic receptor P2RX7 and lead to reduced activity of adenosine 5′-monophosphate–activated protein kinase (AMPK) in HSPC, dramatically increasing mitochondria transfer to BM MSC. Host stromal ME recovery and donor HSPC engraftment were augmented after mitochondria transfer. Deficiency of Cx43 delayed mesenchymal and osteogenic regeneration while in vivo AMPK inhibition increased stromal recovery. As a consequence, the hematopoietic compartment reconstitution was improved because of the recovery of the supportive stromal ME. Our findings demonstrate that healthy donor HSPC not only reconstitute the hematopoietic system after transplantation, but also support and induce the metabolic recovery of their irradiated, damaged ME via mitochondria transfer. Understanding the mechanisms regulating stromal recovery after myeloablative stress are of high clinical interest to optimize BMT procedures and underscore the importance of accessory, non-HSC to accelerate hematopoietic engraftment.
Introduction
Hematopoietic stem cells (HSC) are quiescent cells that reside in the bone marrow (BM) in close proximity to their stromal microenvironment (ME) and continually replenish the circulation with mature cells. The well-established cross talk between HSC and their ME tightly balances their quiescence and self-renewal vs their motility, proliferation, and differentiation upon demand, preserving their repopulation potential.1-3 Lethal or sublethal total body irradiation (TBI) is widely used as a conditioning regimen before hematopoietic stem and progenitor cell (HSPC) transplantation, resulting in the elimination of hematopoiesis and severe damage of the BM ME.4,5
BM HSC transplantation after conditioning is widely used in the clinic to treat pathological mutations in the lymphohematopoietic system.6 Although IV infused BM mesenchymal stromal cells (MSC) cannot engraft into the BM,7 several studies have shown improved efficiency of HSC repopulation and engraftment by BM MSC cotransplant.4,8,9 Higher rates of BM repopulation by LT-HSC in the presence of BM MSC suggest a cross talk between these 2 cell types, which is crucial for successful BM reconstitution. Repopulating HSPC are protected from DNA-damaging agents such as irradiation, by signals from the BM ME, thus explaining the beneficial result of cotransplantation of both populations.5,10 Furthermore, several studies have also demonstrated the facilitating effect of accessory and other dividing hematopoietic progenitors to speed and strengthen the hematopoietic regeneration after irradiation and transplantation.11,12
Quiescent HSC have low reactive oxygen species (ROS) content13-15 which is required to preserve their repopulation potential. In steady-state conditions, HSC ROS levels fluctuate following light/dark signals,16 but regenerative stress upon HSPC cycling induces the accumulation of excess ROS, which may lead to hematopoietic failure in the absence of protecting signals form the BM ME.14 To overcome this insult, HSPC transfer chemotherapy-derived excess ROS to BM MSC in a connexin-43 (Cx43) gap junction–dependent manner.17 Cx43 gap junctions are expressed by both hematopoietic cells and MSC in the BM, and are crucial in the regulation of migration, homing, survival, and senescence of HSPCs.17-21 In the stromal ME, Cx43 was shown to regulate CXCL12 secretion, an essential cytokine which regulates HSPC motility and maintenance.22
BM MSC can metabolically support other cells by transferring their mitochondria, the main ROS-producing organelle in the cell. Mitochondria transfer from BM MSC to epithelial cells protected against acute lung injury,23 and emerging evidence shows that mitochondria transfer from BM MSC to malignant cells as part of their regulatory cross talk provides survival advantage and resistance to chemotherapy.24 Human BM MSC transfer mitochondria to normal or leukemic hematopoietic cells, contributing to their metabolic fitness.25,26 Several mechanisms, among them the mitochondrial ρ-GTPase Miro1,27 PGC-1α,28 and the gap-junction channel Cx43,23 were shown to mediate this transfer via tunneling nanotubes or exosomes29 in a cell-context–dependent manner.
Here we show that TBI, as a preconditioning mechanism before BM transplantation (BMT), damages BM MSC and induces a dysfunction in their mitochondria. Healthy transplanted BM HSPC transfer mitochondria to the damaged MSC, a process requiring the expression of Cx43 and low adenosine 5′-monophosphate–activated protein kinase (AMPK) activity in hematopoietic progenitors. The transferred mitochondria are functional in the recipient MSC and prompt a rapid recovery of the BM ME, improving hematopoietic reconstitution after BMT.
Methods
Mice
Details on the creation of the animals used and subsequent crossings can be found in the supplemental Materials and methods available on the Blood Web site.
Immunofluorescence studies of phenotype, mitochondria transfer, and cell isolation
Immunofluorescence studies were performed by a combination of flow cytometry, sorting, confocal microscopy, and correlative light and electron microscopy (CLEM). Experimental details can be found in the supplemental Materials and methods.
Functional assays
Function analysis was performed by colony-forming-unit, apoptosis analysis, BMT and chimera analysis, mitochondrial transfer, mitochondrial activity, metabolic analysis, and proliferation assays. Experimental details can be found in the supplemental Materials and methods.
Biochemical assays and generation of reconstituted Cx43-expressing HSPC
Biochemical assays were performed by the analysis of glucose uptake and adenosine triphosphate (ATP) levels. Expression of HSPC with full-length Cx43 was performed by retroviral transduction. Experimental details can be found in the supplemental Materials and methods .
Results
Irradiation induces mitochondrial damage in BM stromal ME
To identify the cellular consequences of TBI-induced damage of the BM stromal ME, we first measured the content of MSC in the BM compartment at different time points after TBI and tracked them for 24 hours. The levels of both CD45−/PDGFRα+/Sca-1− (Sca-1− MSC) and CD45−/PDGFRα+/Sca-1+ (Sca-1+ MSC) cells were elevated significantly at 1 hour post-TBI, and gradually decreased in the period of time between 4 and 24 hours post-TBI, suggesting severe damage of both Sca-1− and Sca-1+ BM MSC after irradiation (Figure 1A-B, supplemental Figure 1A). Because mitochondria are the major source of cellular energy and metabolism, we next studied the effect of TBI on the MSC mitochondrial content and function. TBI significantly increased the mitochondrial mass in both Sca-1− and Sca-1+ MSC at 8 hours post-TBI, and were maintained high all through the first 24 hours after irradiation (Figure 1C-D). Similarly, MSC metabolism, assessed as mitochondrial ROS level, was elevated at 24 hours post-TBI in both Sca-1− and Sca-1+ MSC (Figure 1E-F). Mitochondrial dysfunction was confirmed by an analysis of the mitochondrial membrane potential (ΔΨm), which was strongly reduced as early as 8 hours after TBI in both Sca-1− MSC and Sca-1+ MSC (Figure 1G-H). These results imply that TBI leads to severe mitochondrial dysfunction in both Sca-1− and Sca-1+ MSC. To determine whether this mitochondrial dysfunction relates with alterations in upstream glycolysis, we analyzed the cellular uptake of glucose which is upstream to pyruvate and reduced NAD phosphate production and is critical for mitochondrial oxidative phosphorylation, antioxidative potential, and lactate production.30 No sustained difference in glucose uptake was noted in both Sca-1− and Sca-1+ MSC at all the assessed time points post-TBI (Figure 1I-J), suggesting that the irradiation effect has not generated a significant survival bias of MSC with different glucose uptake requirements.31 Other BM niche populations included in the heterogenous pool of CD45−/PDGFRα− cells did not show any major changes in mitochondrial levels (supplemental Figure 1B), albeit their mitochondrial ΔΨm declines dramatically (supplemental Figure 1C) during the first 24 hours after irradiation. Altogether, our results suggest that TBI damages the mitochondria content and function of BM ME cells, albeit with different levels of intensity and time kinetics.
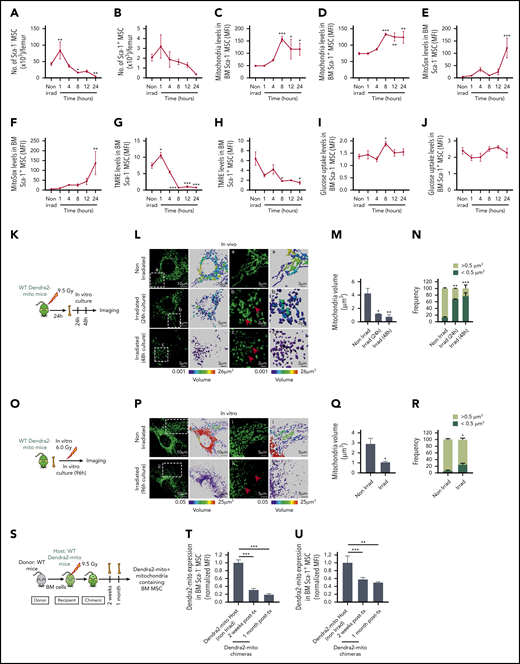
Lethal TBI induces mitochondrial damage in BM stromal precursor cells. (A-J) WT mice were lethally irradiated and Sca-1− (A, C, E, G, I) and Sca-1+ (B, D, F, H, J) BM stromal precursors were analyzed at the indicated time points postirradiation. (A-B) The number of Sca-1− and Sca-1+ BM stromal precursor cells in nonirradiated and irradiated mice. Mitochondrial mass (Mitotracker green staining) (C-D), mitochondrial ROS (MitoSox red staining) levels (E-F), mitochondrial transmembrane potential (TMRE staining) (G-H), and glucose uptake levels (I-J) in BM Sca-1− and Sca-1+ BM stromal precursors before and after irradiation. Data are presented as average of 3 to 7 mice per group. (K) Schematic illustration of in vivo irradiation experiment for stromal mitochondria imaging. (L) Representative confocal microscopy images showing mitochondrial (green) network in MSC cultured for 24 hours and 48 hours after in vivo lethal irradiation. The boxed area (a) and the respective high-magnification images show long, tubular mitochondria network in nonirradiated BM MSC. The boxed areas (b-c) and the respective high-magnification images demonstrate global mitochondrial fragmentation with loss of the long, tubular mitochondrial structures and presence of many small rounded mitochondria at 24 hours and 48 hours (red arrows), respectively. (M) Quantification of mean mitochondrial volume per surface in MSC after in vivo irradiation and culture. (N) Frequency of mitochondrial events with size higher or lower than 0.5 μm3 in volume. (O) Schematic illustration of in vitro irradiation experiment for stromal mitochondria imaging. (P) Representative confocal microscopy images showing mitochondrial (green) network in MSC cultured for 96 hours after in vitro irradiation. The boxed area (i) and the respective high-magnification images show long, tubular mitochondria network in nonirradiated BM MSC (similar to nonirradiated L). The boxed areas (ii) and the respective high-magnification images demonstrate again global mitochondrial fragmentation with loss of the long, tubular mitochondrial structures and presence of many small rounded mitochondria at 96 hours after in vitro irradiation of BM MSC (red arrows). (Q) Quantification of mean mitochondrial volume per surface in MSC after in vitro irradiation. (R) Frequency of mitochondrial events with size higher or lower than 0.5 μm3 in volume. Both in vivo and in vitro experiments were performed as 2 independent experiments. Mitochondrial network and volume were analyzed using Imaris surface building algorithm, and the statistical color coding of mitochondrial volume are shown. Scale bar, 2, 3, 5, and 10 µm. (S-U) Schematic illustration of lethally irradiated Dendra2-mito WT mice transplanted with WT BM cells and analyzed at 2 weeks and 1 month posttransplantation (S). Normalized mean fluorescence intensity level of Dendra2 mitochondria in Sca-1− (T) and Sca-1+ (U) BM stromal precursor cells were analyzed before irradiation, and 2 weeks and 1 month posttransplantation. Data are the average of 3 to 6 mice per group. All data are represented as mean ± SEM. Statistical significance was assessed using 1-way ANOVA except in panels Q and R where the 2-tailed Student t test was used. *P < .05, **P < .01, ***P < .001.
Lethal TBI induces mitochondrial damage in BM stromal precursor cells. (A-J) WT mice were lethally irradiated and Sca-1− (A, C, E, G, I) and Sca-1+ (B, D, F, H, J) BM stromal precursors were analyzed at the indicated time points postirradiation. (A-B) The number of Sca-1− and Sca-1+ BM stromal precursor cells in nonirradiated and irradiated mice. Mitochondrial mass (Mitotracker green staining) (C-D), mitochondrial ROS (MitoSox red staining) levels (E-F), mitochondrial transmembrane potential (TMRE staining) (G-H), and glucose uptake levels (I-J) in BM Sca-1− and Sca-1+ BM stromal precursors before and after irradiation. Data are presented as average of 3 to 7 mice per group. (K) Schematic illustration of in vivo irradiation experiment for stromal mitochondria imaging. (L) Representative confocal microscopy images showing mitochondrial (green) network in MSC cultured for 24 hours and 48 hours after in vivo lethal irradiation. The boxed area (a) and the respective high-magnification images show long, tubular mitochondria network in nonirradiated BM MSC. The boxed areas (b-c) and the respective high-magnification images demonstrate global mitochondrial fragmentation with loss of the long, tubular mitochondrial structures and presence of many small rounded mitochondria at 24 hours and 48 hours (red arrows), respectively. (M) Quantification of mean mitochondrial volume per surface in MSC after in vivo irradiation and culture. (N) Frequency of mitochondrial events with size higher or lower than 0.5 μm3 in volume. (O) Schematic illustration of in vitro irradiation experiment for stromal mitochondria imaging. (P) Representative confocal microscopy images showing mitochondrial (green) network in MSC cultured for 96 hours after in vitro irradiation. The boxed area (i) and the respective high-magnification images show long, tubular mitochondria network in nonirradiated BM MSC (similar to nonirradiated L). The boxed areas (ii) and the respective high-magnification images demonstrate again global mitochondrial fragmentation with loss of the long, tubular mitochondrial structures and presence of many small rounded mitochondria at 96 hours after in vitro irradiation of BM MSC (red arrows). (Q) Quantification of mean mitochondrial volume per surface in MSC after in vitro irradiation. (R) Frequency of mitochondrial events with size higher or lower than 0.5 μm3 in volume. Both in vivo and in vitro experiments were performed as 2 independent experiments. Mitochondrial network and volume were analyzed using Imaris surface building algorithm, and the statistical color coding of mitochondrial volume are shown. Scale bar, 2, 3, 5, and 10 µm. (S-U) Schematic illustration of lethally irradiated Dendra2-mito WT mice transplanted with WT BM cells and analyzed at 2 weeks and 1 month posttransplantation (S). Normalized mean fluorescence intensity level of Dendra2 mitochondria in Sca-1− (T) and Sca-1+ (U) BM stromal precursor cells were analyzed before irradiation, and 2 weeks and 1 month posttransplantation. Data are the average of 3 to 6 mice per group. All data are represented as mean ± SEM. Statistical significance was assessed using 1-way ANOVA except in panels Q and R where the 2-tailed Student t test was used. *P < .05, **P < .01, ***P < .001.
To further explore the effect of TBI on MSC mitochondrial morphology, we used Dendra2-mitochondria (Dendra2-mito)–transgenic mice in which the Dendra2 fluorescent protein is fused with the targeting signal of subunit 8a of mitochondrial cytochrome oxidase (Cox8a),32 a mitochondrial transmembrane protein, and allows measurements of mitochondrial mass.33 These mice were either lethally irradiated, and their BM CD45− MSC were tested for the mitochondria networking structure, or primary MSC were isolated from these mice and irradiated in vitro to test the consequences of TBI on the mitochondrial structure. Confocal microscopy of Dendra2-mito structure in in vivo– or in vitro–irradiated BM MSC demonstrates global mitochondrial fragmentation with the presence of many small, round/ovoid-shaped mitochondria, resulting in reduced mitochondrial volume as compared with nonirradiated counterparts showing tubular mitochondrial networks (Figure 1K-R; supplemental Videos 1-3). In contrast, TBI exerted a minimal effect on the mitochondrial networking of another functionally relevant ME cell population like BM endothelial cells (Lin−/CD45−/Sca-1+/CD31+) (supplemental Figure 1D-H), and did not change their overall mitochondrial mass (supplemental Figure 1I).
Further, to explore the long-term effects of TBI on MSC mitochondria content, Dendra2-mito mice were lethally irradiated and transplanted with wild-type (WT) HSPC, and the level of Dendra2+ mitochondria in BM MSC was assessed at 2 weeks and 1 month post-TBI (Figure 1S). A strong reduction in BM stromal cell mitochondria levels was observed in Sca-1− MSC, albeit at a lower extent in Sca-1+ MSC at 2 weeks and 1 month post-TBI (Figure 1T-U), suggesting that both populations of MSC, and especially Sca-1− MSC, have a decrease in their content of host transgenic mitochondria. Overall, our data demonstrate that TBI induced damage to BM MSC, leading to mitochondria dysfunction that is mostly prominent in Sca-1− MSC.
Donor hematopoietic cells transfer functional mitochondria to BM MSC
Since TBI preconditioning for BMT damages the MSC by affecting their mitochondria function, we hypothesized that healthy HSPC after transplantation transfer some of their mitochondria to the BM MSC in order to maintain their function and increase their recovery. To test this hypothesis, we created chimeric mice by transplanting Dendra2-mito CD45+ cells into lethally irradiated WT recipients (Figure 2A). At 2 weeks and again at 1-month posttransplantation, Dendra2-mito levels in recipient WT MSC were examined and found 89 ± 1.3% of these cells to express donor-derived Dendra2+ mitochondria (Figure 2B-C; supplemental Figure 2A). These results suggest that mitochondria are indeed transferred from donor hematopoietic cells to the BM stromal ME after transplantation. Next, we asked whether mitochondria can be transferred in the opposite direction and thus chimeric mice were established by transplanting donor WT CD45+ cells into lethally irradiated Dendra2-mito recipient mice (Figure 2D). After 2 weeks and 1 month from transplantation, 18 ± 2.5% of WT HSC (Lin−/CD34−/c-Kit+/Sca-1+) expressed Dendra2+ mitochondria (Figure 2E-F; supplemental Figure 2B), suggesting that mitochondria can be transferred also from the BM MSC to the hematopoietic cells after transplantation, but to a much lower extent. The transfer of mitochondria from donor BM leukocytes to BM MSC was also confirmed in an isolated in vitro system after overnight coculture of primary BM MSC with BM CD45+ Dendra2-mito cells. We observed 21 ± 1.3% of the BM MSC to contain donor-derived Dendra2-mito in in vitro cocultures, further confirming our in vivo findings (Figure 2G-H). As a consequence, the mitochondrial mass of recipient BM MSC was increased (approximately twofold) after coculture with BM HSPC (Figure 2I). Transfer of mitochondria from donor BM leukocytes to BM MSC was also tracked by time-lapse confocal microscopy in cocultures of CD45+ Dendra2-mito cells with primary BM MSC (Figure 2J). Transfer of Dendra2 mitochondria from donor-derived BM leukocytes to BM MSC starts soon after the coculture is established (2-3 hours), and both orthogonal views and 3-dimensional reconstitution images clearly demonstrate the presence of donor-derived Dendra2-mito in the recipient BM MSC (supplemental Figure 2C-D and supplemental Video 4). Similar levels of mitochondrial transfer from human BM CD34+ cells to murine BM stroma was evident by analyzing the presence of human mitochondrial DNA (Mitotracker Deep Red+/Dendra2− mitochondria) in 20% of Dendra2-mito BM MSC after coculture at a ratio of 5 CD34+ cells per MSC (supplemental Figure 3A-B).
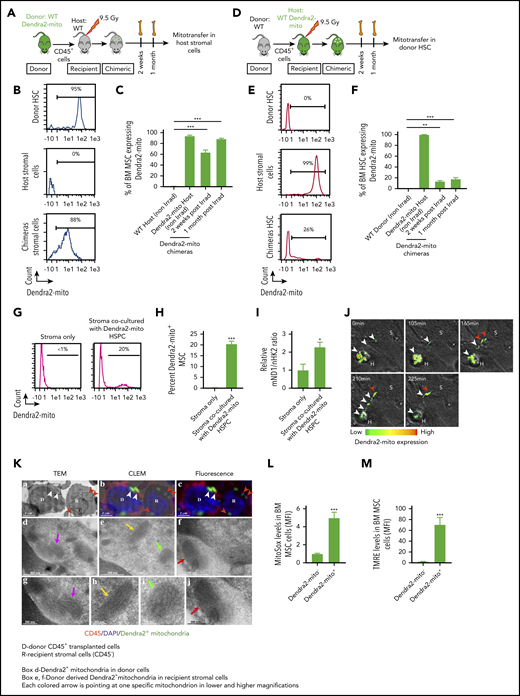
Donor hematopoietic cells transfer functional mitochondria to the irradiated host BM MSC following total body irradiation. (A-C) Schematic illustration of transplantation protocol. Lethally irradiated congenic WT mice transplanted with CD45+ BM cells obtained from Dendra2-mito WT mice and analyzed 2 weeks and 1 month posttransplantation (A). Representative histograms after 1 month posttransplantation (B) and quantified analyses (C) show the levels of Dendra2+ mitochondria transfer from donor HSPC to host BM-MSC (CD45−/PDGFRα+/Sca-1−). Data are the average of 3 to 5 mice per group. (D-F) Lethally irradiated WT Dendra2-mito mice transplanted with CD45+ BM cells obtained from congenic WT mice and analyzed 2 weeks and 1 month posttransplantation (D). Representative histograms after 1 month posttransplantation (E) and quantified summary (F) show the levels of Dendra2+ mitochondria transfer from host Dendra2-mito stromal cells to WT donor HSC (CD34−/Lin−/Sca-1+/c-Kit+). Data are the average of 3 to 6 mice per group. (G-I) BM MSC were cocultured with CD45+ cells isolated from Dendra2-mito WT mice for 16 hours, and the transfer of mitochondria from Dendra2-mito CD45+ cells to MSC was analyzed. Histograms (G) and bar diagram (H) representing the percentage of MSC containing donor-derived Dendra2+ mitochondria. (I) Relative quantification of mitochondrial content in stromal cells cocultured with or without Dendra2-mito CD45+ cells was analyzed by real-time polymerase chain reaction using ND1 gene belonging to mitochondrial DNA (mND1) and nuclear hexokinase 2 (nHK2) gene. n = 4-6 independent experiments. (J) Representative example of mitochondrial transfer kinetics followed for up to 225 min. Confocal spinning disk microscopy of hematopoietic Dendra2-mito cells cocultured with WT stromal precursors. Heat map shows the Dendra2 signal intensity. Images taken at indicated time points show the transfer of mitochondria from 1 hematopoietic cell (H) toward a neighboring stromal cell (S) after a long, thin hematopoietic cell extension. White arrows depict mitochondria moving away from H to S and red arrows depict mitochondria already transferred to S. (K) TEM images of mitochondria transfer in an in vivo setting. WT Dendra2-mito CD45+ cells were transplanted in lethally irradiated WT congenic mice and the transfer of Dendra2+ mitochondria from HSPC to MSC was analyzed 4 months posttransplantation. (a) Representative TEM images showing donor (D) and recipient (R) cells, and mitochondrial cristae. (b) Overlay of TEM image with identical fluorescent micrograph (correlative light electron microscopy, CLEM). (c) Fluorescent microscopy image showing donor CD45+ hematopoietic cells (red) and Dendra2+ mitochondria (green). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) . The short white arrows in panels a-c show Dendra2+ mitochondria in donor CD45+ cells. The short red arrows in panel a-c represent donor Dendra2+ mitochondria in recipient stromal cells. The boxed areas (d, e, and f) in panel a are magnified in TEM image panels d, e and f, respectively. TEM magnification of mitochondria indicated by a long magenta arrow in panel d is presented in TEM micrograph (g). TEM magnification of mitochondria indicated by a long yellow arrow and a green arrow in panel e are presented in TEM micrographs panels h and i, respectively. TEM magnification of mitochondria indicated by a long red arrow in panel f is presented in TEM micrograph (j). All these mitochondrial images represent donor-derived Dendra2+ mitochondria which either persist in the hematopoietic (CD45+, red fluorescent) donor cell (d, g) or have been transferred to a recipient BM stromal cell (CD45−, with no red fluorescence; in e, f, h, i, and j). TEM micrographs in panels g, h, i, and j provide morphological detail on mitochondrial cristae and membranes. Scale bar, 2 µm, 500 nm, and 200 nm. (L-M) Mitochondrial ROS levels (L) and (M) membrane potential in host MSC containing (Dendra2-mito+) or not (Dendra2-mito−) donor-derived Dendra2+ mitochondria assessed at 1 month posttransplantation (n = 5 mice per group). All data are presented as mean ± SEM. Statistical significance was assessed using 2-tailed Student t test except in panels C and F, where one-way ANOVA was used. **P < .01, ***P < .001.
Donor hematopoietic cells transfer functional mitochondria to the irradiated host BM MSC following total body irradiation. (A-C) Schematic illustration of transplantation protocol. Lethally irradiated congenic WT mice transplanted with CD45+ BM cells obtained from Dendra2-mito WT mice and analyzed 2 weeks and 1 month posttransplantation (A). Representative histograms after 1 month posttransplantation (B) and quantified analyses (C) show the levels of Dendra2+ mitochondria transfer from donor HSPC to host BM-MSC (CD45−/PDGFRα+/Sca-1−). Data are the average of 3 to 5 mice per group. (D-F) Lethally irradiated WT Dendra2-mito mice transplanted with CD45+ BM cells obtained from congenic WT mice and analyzed 2 weeks and 1 month posttransplantation (D). Representative histograms after 1 month posttransplantation (E) and quantified summary (F) show the levels of Dendra2+ mitochondria transfer from host Dendra2-mito stromal cells to WT donor HSC (CD34−/Lin−/Sca-1+/c-Kit+). Data are the average of 3 to 6 mice per group. (G-I) BM MSC were cocultured with CD45+ cells isolated from Dendra2-mito WT mice for 16 hours, and the transfer of mitochondria from Dendra2-mito CD45+ cells to MSC was analyzed. Histograms (G) and bar diagram (H) representing the percentage of MSC containing donor-derived Dendra2+ mitochondria. (I) Relative quantification of mitochondrial content in stromal cells cocultured with or without Dendra2-mito CD45+ cells was analyzed by real-time polymerase chain reaction using ND1 gene belonging to mitochondrial DNA (mND1) and nuclear hexokinase 2 (nHK2) gene. n = 4-6 independent experiments. (J) Representative example of mitochondrial transfer kinetics followed for up to 225 min. Confocal spinning disk microscopy of hematopoietic Dendra2-mito cells cocultured with WT stromal precursors. Heat map shows the Dendra2 signal intensity. Images taken at indicated time points show the transfer of mitochondria from 1 hematopoietic cell (H) toward a neighboring stromal cell (S) after a long, thin hematopoietic cell extension. White arrows depict mitochondria moving away from H to S and red arrows depict mitochondria already transferred to S. (K) TEM images of mitochondria transfer in an in vivo setting. WT Dendra2-mito CD45+ cells were transplanted in lethally irradiated WT congenic mice and the transfer of Dendra2+ mitochondria from HSPC to MSC was analyzed 4 months posttransplantation. (a) Representative TEM images showing donor (D) and recipient (R) cells, and mitochondrial cristae. (b) Overlay of TEM image with identical fluorescent micrograph (correlative light electron microscopy, CLEM). (c) Fluorescent microscopy image showing donor CD45+ hematopoietic cells (red) and Dendra2+ mitochondria (green). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) . The short white arrows in panels a-c show Dendra2+ mitochondria in donor CD45+ cells. The short red arrows in panel a-c represent donor Dendra2+ mitochondria in recipient stromal cells. The boxed areas (d, e, and f) in panel a are magnified in TEM image panels d, e and f, respectively. TEM magnification of mitochondria indicated by a long magenta arrow in panel d is presented in TEM micrograph (g). TEM magnification of mitochondria indicated by a long yellow arrow and a green arrow in panel e are presented in TEM micrographs panels h and i, respectively. TEM magnification of mitochondria indicated by a long red arrow in panel f is presented in TEM micrograph (j). All these mitochondrial images represent donor-derived Dendra2+ mitochondria which either persist in the hematopoietic (CD45+, red fluorescent) donor cell (d, g) or have been transferred to a recipient BM stromal cell (CD45−, with no red fluorescence; in e, f, h, i, and j). TEM micrographs in panels g, h, i, and j provide morphological detail on mitochondrial cristae and membranes. Scale bar, 2 µm, 500 nm, and 200 nm. (L-M) Mitochondrial ROS levels (L) and (M) membrane potential in host MSC containing (Dendra2-mito+) or not (Dendra2-mito−) donor-derived Dendra2+ mitochondria assessed at 1 month posttransplantation (n = 5 mice per group). All data are presented as mean ± SEM. Statistical significance was assessed using 2-tailed Student t test except in panels C and F, where one-way ANOVA was used. **P < .01, ***P < .001.
Next, to track the transfer of Dendra2 mitochondria from donor BM leukocytes to BM MSC in an in vivo setting, and to examine whether the transferred Dendra2 mitochondria retain their morphology in the recipient MSC, we transplanted Dendra2-mito CD45+ cells into lethally irradiated WT recipients and applied a high-resolution correlative light and electron microscopy (CLEM) to access the acquired Dendra2 mitochondria structure in the MSC. As shown in Figure 2K, recipient BM MSC contain donor-derived Dendra2-mito which overlap with a mitochondria structure by TEM, confirming that, indeed, the transferred Dendra2-mito is part of a true mitochondria, and it retains its morphology after transfer. Finally, we wanted to know whether the mitochondria that are transferred from hematopoietic cells to BM MSC are indeed functional. Therefore, mitochondrial ROS levels, as well as ΔΨM, were measured in MSC of chimeric mice 1 month after BMT of Dendra2-mito CD45+ cells into WT recipients. Recipient BM MSC that contained donor-derived Dendra2 mitochondria demonstrated increased ROS production and higher ΔΨM as compared with recipient BM MSC with no donor-derived Dendra2 mitochondria, suggesting that the mitochondria transferred from donor BM leukocytes to BM MSC are metabolically active and significantly reprogram the overall mitochondrial metabolism of the recipient BM MSC (Figure 2L-M).
Deficiency of Cx43 in HSPC decreased their ability to transfer mitochondria to BM Sca-1− MSC cells and not to other BM stromal populations
Organelle exchange between cells is either cell-contact dependent34 or occurs through their secretion in extracellular vesicles.23 To identify the mode of mitochondria transfer between HSPC and BM MSC, we first established in vitro cocultures of Dendra2-mito CD45+ cells and WT primary MSC with a mechanical barrier between the 2 cell types. No Dendra2+ mitochondria transfer from HSPC to BM MSC was found in the noncontacting cocultures (Figure 3A), suggesting that HSPC and MSC need a physical contact to transfer mitochondria. A major contributor to cell contact in the BM is Cx43 gap junction, which was previously shown to regulate HSPC motility and survival as well as stromal secretion of CXCL12.22 In addition, HSPC Cx43 plays a protective role during myeloablative conditions, facilitating the transfer of potentially lethal ROS from HSPC to the BM MSC and preventing ROS-mediated HSPC damage.17 Therefore, the role of Cx43 in cell-contact–dependent mitochondria transfer was examined. Time-dependent analysis of mitochondria uptake by MSC in an in vitro coculture of WT or Cx43 deficient (Cx43Δ/Δ) Dendra2-mito HSPC with primary WT MSC show reduced transfer of mitochondria from Cx43Δ/Δ HSPC to BM stroma at 16 hours (Figure 3B; supplemental Figure 4A-C). Similarly, both mitochondrial ROS levels and the ΔΨM were lower in BM MSC containing donor-derived mitochondria that were cocultured with Dendra2-mito Cx43Δ/Δ HSPC as compared with WT HSPC (Figures 3C-D; supplemental Figure 4D-E). This correlates with the decreased mitochondrial transfer occurring in cocultures of Cx43-deficient HSPC with WT stroma. Transfecting Cx43Δ/Δ HSPC with a vector expressing WT FL-Cx43 followed by in vitro coculture with WT primary MSC restored mitochondria transfer to the levels seen for WT HSPC transfer, further confirming the requirement of Cx43 in HSPCs for mitochondria transfer (Figure 3E). To further identify the role of hematopoietic Cx43 in mitochondrial transfer in an in vivo setting, we established chimeric mice by transplanting Lin−/CD51−/Dendra2-mito WT or Cx43Δ/Δ hematopoietic progenitors into lethally irradiated WT recipients, and the level of mitochondria transferred from donor HSPC to recipient mice MSC was analyzed at different times posttransplantation (Figure 3F). Hematopoietic Cx43 deficiency significantly attenuated the transfer of healthy mitochondria from HSPC to BM Sca-1− MSC (with 50% reduction in mitochondria transfer to Sca-1− MSC) at all the time points analyzed post-TBI (Figure 3G-H; supplemental Figure 5A) in comparison with WT HSPC mitochondria transfer. Interestingly, the deficiency of Cx43 in HSPC did not modify the transfer of mitochondria to other BM stromal populations (supplemental Figure 5A-E). Mitochondria transfer from Col-1cre–expressing/Cx43-deficient MSC20 to HSC was also unaltered (supplemental Figure 4F-J). These data indicate that hematopoietic Cx43 exquisitely mediates mitochondria transfer from healthy donor HSPC to damaged, recipient BM Sca-1− MSC in vivo. Interestingly, the reduced mitochondria transfer from Cx43Δ/Δ HSPC to stromal cells, affected only BM Sca-1− MSC and not other stromal populations, as shown by their reduced overall mitochondria ROS levels (Figure 3I; supplemental Figure 5F-I) as well as diminished ΔΨM (Figure 3J; supplemental Figure 5J-M). These results suggest that either fewer mitochondria were transferred to BM stromal recipient cells from Cx43Δ/Δ hematopoietic progenitors or that the mitochondria transferred from Cx43Δ/Δ hematopoietic progenitors were dysfunctional. To address this question, we isolated functional mitochondria from Dendra2-mito Lin− WT and Cx43Δ/Δ cells, incubated them with irradiated WT primary MSC for 24 hours and 48 hours, and found that stromal MSC incorporated isolated mitochondria at an equal level and with similar levels of ROS production capacity regardless of the mitochondria cellular origin (Figure 3K-M; supplemental Figure 4K-M). To gain insight whether transfer of mitochondria improves MSC oxidative phosphorylation, the mitochondrial oxygen consumption rate (OCR) in MSC was analyzed in an in vivo and in vitro setting. The level of maximal mitochondrial OCR, spare respiratory capacity, and bioenergetic health index was significantly lower in BM stromal cells obtained from H-Cx43Δ/Δ chimeric mice (Figure 3N-O). In contrast, equal increase in mitochondrial OCR was observed in stromal cells cocultured with isolated functional mitochondria from Dendra2-mito WT and Cx43Δ/Δ HSPC (supplemental Figure 4N-O). Altogether, these results strongly suggest that cellular contact between HSPC and BM MSC is indispensable for functional mitochondria transfer and the reduced transfer of mitochondria from Cx43Δ/Δ HSPC results in overall diminished mitochondrial activity in the host BM MSC.
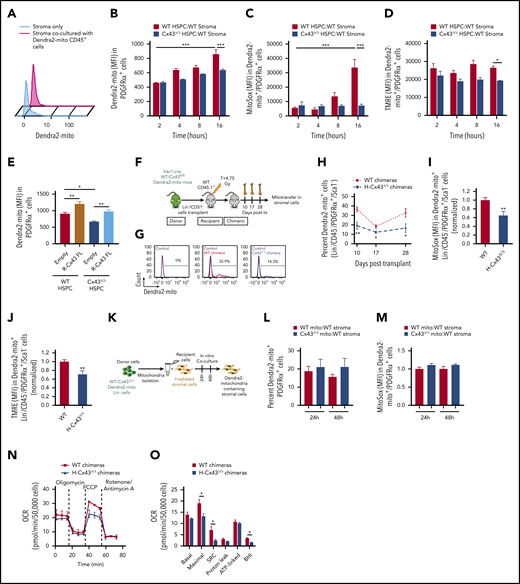
Mitochondrial transfer from Connexin 43 deficient donor HSPC to BM MSC is decreased. (A) Level of Dendra2-mito transfer from HSPC to BM stromal cells in coculture without contact in trans-wells of 0.3µm. (B-D) WT or H-Cx43Δ/Δ Dendra2-mito Lin-negative cells were cocultured on WT MSC and the transfer of Dendra2+ mitochondria in MSC was analyzed. Mean fluorescence intensity of Dendra2 in PDGFRα+ MSC after 2, 4, 8, and 16 hours of coculture (B). Mitochondrial ROS levels (C) and ΔΨm (D) in PDGFRα+ MSC containing donor-derived Dendra2+ mitochondria at different times of coculture. Data are the average of 3 to 6 independent experiments. (E) Lin-negative cells from WT or Vav1-cre Cx43fl/fl Dendra2-mito mice were transduced with empty or Cx43-full length (R-Cx43-FL) retrovirus vector, followed by coculture over WT stroma for 16 hours. Overexpression of R-Cx43-FL in Cx43Δ/Δ Dendra2+ HSPC rescue mitochondrial transfer in PDGFRα+ MSC. The frequency of mitochondria transfer in PDGFRα+ MSC was also increased in R-Cx43-FL transduced WT HSPC. Data are the average of 3 independent experiments. (F-J) Schematic illustration of lethally irradiated congenic WT CD45.1+ mice transplanted with WT or Cx43Δ/Δ Dendra2-mito Lin−/CD51− cells and analyzed at days 10, 17, and 28 posttransplantation (F). Representative histograms (G) and bar diagram (H) show the frequency of BM Lin−/CD45−/PDGFRα+/Sca-1− MSC containing Dendra2+ mitochondria from donor hematopoiesis at the indicated days posttransplantation. Mitochondrial ROS levels (I), and ΔΨm (J) in WT and H-Cx43Δ/Δ chimeric mice BM Lin−/CD45−/PDGFRα+/Sca-1− MSC containing donor-derived Dendra2+ mitochondria after 28 days posttransplantation are shown. (n = 4-9 mice per group, 2 independent experiments). (K-M) Incorporation of isolated mitochondria from HSPC is independent of the expression of Cx43 in source HSPC. Dendra2+ mitochondria were isolated from WT and Cx43Δ/Δ Dendra2-mito Lin-negative cells and coculture over-irradiated (7.5 Gy) WT primary stroma for 24 and 48 hours (K). Bar graphs show the frequencies of PDGFRα+ MSC containing extracellular Dendra2+ mitochondria at indicated time points (L), and mitochondrial ROS production in PDGFRα+ MSC containing extracellular Dendra2+ mitochondria (M). Data are the average of 3 to 5 independent experiments. Dendra2+ mitochondria isolated from WT HSPC (WT mito). Dendra2+ mitochondria isolated from Cx43Δ/Δ HSPC (Cx43Δ/Δ mito). (N-O) BM Lin−/CD45− cells containing donor-derived Dendra2+ mitochondrial were sorted from WT and H-CX43Δ/Δ chimeric mice (1 month posttransplantation) and mitochondrial OCR was measured by Seahorse XFe96-Analyzer using sequential injections of oligomycin, FCCP, and Rotenone (N). Quantification summary of mitochondrial OCR in WT and H-CX43Δ/Δ chimeric mice Lin−/CD45− cells containing donor-derived mitochondria (data are the average of 3 independent experiments with 2 to 4 technical replicates) (O). All data represented as mean ± SEM. Statistical significance was assessed using 1-way ANOVA except in panels H, I, J, and O where 2-tailed Student t tests were used. *P < .05, **P < .01, ***P < .001. BHI, bioenergetic health index; FCCP, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; SRC, spare respiratory capacity.
Mitochondrial transfer from Connexin 43 deficient donor HSPC to BM MSC is decreased. (A) Level of Dendra2-mito transfer from HSPC to BM stromal cells in coculture without contact in trans-wells of 0.3µm. (B-D) WT or H-Cx43Δ/Δ Dendra2-mito Lin-negative cells were cocultured on WT MSC and the transfer of Dendra2+ mitochondria in MSC was analyzed. Mean fluorescence intensity of Dendra2 in PDGFRα+ MSC after 2, 4, 8, and 16 hours of coculture (B). Mitochondrial ROS levels (C) and ΔΨm (D) in PDGFRα+ MSC containing donor-derived Dendra2+ mitochondria at different times of coculture. Data are the average of 3 to 6 independent experiments. (E) Lin-negative cells from WT or Vav1-cre Cx43fl/fl Dendra2-mito mice were transduced with empty or Cx43-full length (R-Cx43-FL) retrovirus vector, followed by coculture over WT stroma for 16 hours. Overexpression of R-Cx43-FL in Cx43Δ/Δ Dendra2+ HSPC rescue mitochondrial transfer in PDGFRα+ MSC. The frequency of mitochondria transfer in PDGFRα+ MSC was also increased in R-Cx43-FL transduced WT HSPC. Data are the average of 3 independent experiments. (F-J) Schematic illustration of lethally irradiated congenic WT CD45.1+ mice transplanted with WT or Cx43Δ/Δ Dendra2-mito Lin−/CD51− cells and analyzed at days 10, 17, and 28 posttransplantation (F). Representative histograms (G) and bar diagram (H) show the frequency of BM Lin−/CD45−/PDGFRα+/Sca-1− MSC containing Dendra2+ mitochondria from donor hematopoiesis at the indicated days posttransplantation. Mitochondrial ROS levels (I), and ΔΨm (J) in WT and H-Cx43Δ/Δ chimeric mice BM Lin−/CD45−/PDGFRα+/Sca-1− MSC containing donor-derived Dendra2+ mitochondria after 28 days posttransplantation are shown. (n = 4-9 mice per group, 2 independent experiments). (K-M) Incorporation of isolated mitochondria from HSPC is independent of the expression of Cx43 in source HSPC. Dendra2+ mitochondria were isolated from WT and Cx43Δ/Δ Dendra2-mito Lin-negative cells and coculture over-irradiated (7.5 Gy) WT primary stroma for 24 and 48 hours (K). Bar graphs show the frequencies of PDGFRα+ MSC containing extracellular Dendra2+ mitochondria at indicated time points (L), and mitochondrial ROS production in PDGFRα+ MSC containing extracellular Dendra2+ mitochondria (M). Data are the average of 3 to 5 independent experiments. Dendra2+ mitochondria isolated from WT HSPC (WT mito). Dendra2+ mitochondria isolated from Cx43Δ/Δ HSPC (Cx43Δ/Δ mito). (N-O) BM Lin−/CD45− cells containing donor-derived Dendra2+ mitochondrial were sorted from WT and H-CX43Δ/Δ chimeric mice (1 month posttransplantation) and mitochondrial OCR was measured by Seahorse XFe96-Analyzer using sequential injections of oligomycin, FCCP, and Rotenone (N). Quantification summary of mitochondrial OCR in WT and H-CX43Δ/Δ chimeric mice Lin−/CD45− cells containing donor-derived mitochondria (data are the average of 3 independent experiments with 2 to 4 technical replicates) (O). All data represented as mean ± SEM. Statistical significance was assessed using 1-way ANOVA except in panels H, I, J, and O where 2-tailed Student t tests were used. *P < .05, **P < .01, ***P < .001. BHI, bioenergetic health index; FCCP, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; SRC, spare respiratory capacity.
AMPK in HSPC controls mitochondrial transfer to BM MSC
To show a connection between mitochondria transfer and irradiation-induced damage to the stromal ME, we irradiated explanted WT primary BM MSC at doses of 9.5 and 20 Gy which were expected to induce death of ∼90% and >99% of mesenchymal progenitors, respectively.35 At 48 hours postirradiation, we cocultured the MSC with BM CD45+ Dendra2-mito cells. As anticipated, the level of mitochondrial transfer to irradiated MSC was higher than to nonirradiated MSC (Figure 4A), suggesting that mitochondria transfer from HSPC is augmented by irradiation-induced damage of the stromal compartment. The mitochondria are the main metabolic sites in the cell,36 and as such they tightly regulate cellular ATP levels. To examine whether intracellular ATP levels affect mitochondria transfer, we pretreated either WT MSC or isolated CD45+ Dendra2-mito cells with the glycolysis enhancer buffer Rejuvesol,37 a nucleoside-containing solution that induced strong ATP production in BM MSC and only modestly in cycling hematopoietic progenitors (supplemental Figure 6A-B). After drug removal, these cells were cocultured with untreated CD45+ Dendra2-mito or WT MSC, respectively, and mitochondria transfer was measured. Only pretreatment of the hematopoietic cells resulted in increased mitochondria transfer (Figure 4B), suggesting that the elevated intracellular ATP concentration in hematopoietic cells can stimulate the transfer of mitochondria from HSPC to BM MSC. To further examine the role of ATP in mitochondria transfer, we cocultured isolated CD45+ Dendra2-mito cells with WT BM MSC together with Rejuvesol and a specific inhibitor of the ATP receptor, P2RX7. High mitochondria transfer induced by Rejuvesol was 50% reduced upon cotreatment with the P2RX7 receptor inhibitor (Figure 4C), implying that high ATP levels induced signaling and stimulated mitochondria transfer. A well-known sensor of ATP levels in the cell is AMPK.38 Therefore, we tested whether AMPK plays a role in mitochondria transfer by treating the coculture of Dendra2-mito hematopoietic cells and WT MSC with the AMPK inhibitor, BML-275 (compound C), and the AMPK activator, AICAR (5-aminoimidazole-4-carboxamide ribonucleotide). AMPK inhibition elevated in vitro mitochondria transfer from hematopoietic cells to MSC, whereas AMPK activation reduced this transfer (Figure 4D-E). To further explore which cellular compartment is sensitive to AMPK inhibition–mediated mitochondria transfer, we pretreated either WT MSC or isolated CD45+ Dendra2-mito cells with the AMPK inhibitor, and after drug removal cocultured these cells with untreated CD45+ Dendra2-mito or WT MSC, respectively. Only pretreatment of the hematopoietic cells with the AMPK inhibitor resulted in increased mitochondria transfer from HSPC to BM MSC (Figure 4F), suggesting that AMPK activity in the hematopoietic compartment is a critical regulator of mitochondria transfer. Altogether, these results suggest that elevated ATP levels in hematopoietic cells signal via hematopoietic P2RX7 receptor as well as inhibit AMPK activation in these cells, all leading to stimulation of mitochondria transfer to the stromal ME. To further test this hypothesis, we cocultured isolated CD45+ Dendra2-mito cells with WT MSC together with AMPK inhibitor and Rejuvesol and found an additive effect, whereas treatment with both AMPK and P2RX7 receptor inhibitors did not significantly reduce the high mitochondria transfer induced by AMPK inhibitor only (Figure 4G). These results suggest that AMPK is a downstream regulator of mitochondria transfer in HSPC dependent on P2RX7 signaling. Finally, we examined the effect of AMPK inhibition on mitochondria transfer in vivo. AMPK inhibition increased mitochondria transfer regardless of the origin of the hematopoietic cells (WT or Cx43Δ/Δ BM) (Figure 4H-I), suggesting that the activity of AMPK is downstream to Cx43 or represses alternative pathways of mitochondrial transfer.

AMPK inhibition in HSPC controls mitochondria transfer to BM MSC. (A) Primary BM MSC were irradiated (9.5 Gy or 20 Gy), and 48 hours postirradiation, were cocultured with CD45+ cells isolated from Dendra2-mito WT mice for 16 h at 37°C, and the transfer of mitochondria in MSC was analyzed. (B) Primary BM MSC or CD45+ cells isolated from Dendra2-mito WT mice were pretreated with Rejuvesol for 4 hours and washed with intact media. Rejuvesol-pretreated stromal or CD45+ cells were cocultured with CD45+ cells isolated from Dendra2-mito WT mice or WT primary MSC, respectively, for 16 hours, and the transfer of mitochondria in MSC was analyzed. (C) Primary BM MSC were cocultured with CD45+ cells isolated from Dendra2-mito WT mice with/without Rejuvesol and P2RX7 receptor inhibitor for 16 hours and the transfer of mitochondria in MSC was analyzed. (D-E) Primary BM MSC were cocultured with CD45+ cells isolated from Dendra2-mito WT mice with/without AMPK inhibitor (BML-275) or AMPK activator (AICAR) for 16 hours, and the transfer of mitochondria in MSC was analyzed. Representative plots (D) and quantification summary (E) shows the effect of AMPK inhibitor and activator on mitochondria transfer from HSPC to BM MSC. (F) Primary BM MSC or CD45+ cells isolated from Dendra2-mito WT mice were pretreated with AMPK inhibitor for 4 hours and washed with intact media. AMPK inhibitor–pretreated stromal and CD45+ cells were cocultured with CD45+ cells isolated from Dendra2-mito WT mice or WT stromal cells, respectively, for 16 hours and the transfer of mitochondria in MSC was analyzed. (G) Primary BM MSC were cocultured with CD45+ cells isolated from Dendra2-mito WT mice with/without AMPK inhibitor, P2RX7 receptor inhibitor and Rejuvesol for 16 hours, and the transfer of mitochondria in MSC was analyzed. (H-I) Schematic illustration of lethally irradiated congenic WT CD45.1+ mice transplanted with WT or Cx43Δ/Δ Dendra2-mito HSPC and treated with vehicle control or AMPK inhibitor (BML-275, 10 mg/kg, interperitoneally) on days 5, 6, and 7 posttransplantation, and analyzed 2.5 hours after the last dose (H). Transfer of Dendra2+ mitochondria from donor HSPC to BM Lin−/CD45−/PDGFRα+/Sca-1− cells in WT and H-Cx43Δ/Δ chimeric mice treated with vehicle control or AMPK inhibitor (I). n = 8 mice per group, 2 independent experiments. All in vitro data are the average of 4 to 6 independent experiments. Data represented as mean ± SEM and statistical significance was assessed using 1-way ANOVA. *P < .05, **P < .01 and ***P < .001.
AMPK inhibition in HSPC controls mitochondria transfer to BM MSC. (A) Primary BM MSC were irradiated (9.5 Gy or 20 Gy), and 48 hours postirradiation, were cocultured with CD45+ cells isolated from Dendra2-mito WT mice for 16 h at 37°C, and the transfer of mitochondria in MSC was analyzed. (B) Primary BM MSC or CD45+ cells isolated from Dendra2-mito WT mice were pretreated with Rejuvesol for 4 hours and washed with intact media. Rejuvesol-pretreated stromal or CD45+ cells were cocultured with CD45+ cells isolated from Dendra2-mito WT mice or WT primary MSC, respectively, for 16 hours, and the transfer of mitochondria in MSC was analyzed. (C) Primary BM MSC were cocultured with CD45+ cells isolated from Dendra2-mito WT mice with/without Rejuvesol and P2RX7 receptor inhibitor for 16 hours and the transfer of mitochondria in MSC was analyzed. (D-E) Primary BM MSC were cocultured with CD45+ cells isolated from Dendra2-mito WT mice with/without AMPK inhibitor (BML-275) or AMPK activator (AICAR) for 16 hours, and the transfer of mitochondria in MSC was analyzed. Representative plots (D) and quantification summary (E) shows the effect of AMPK inhibitor and activator on mitochondria transfer from HSPC to BM MSC. (F) Primary BM MSC or CD45+ cells isolated from Dendra2-mito WT mice were pretreated with AMPK inhibitor for 4 hours and washed with intact media. AMPK inhibitor–pretreated stromal and CD45+ cells were cocultured with CD45+ cells isolated from Dendra2-mito WT mice or WT stromal cells, respectively, for 16 hours and the transfer of mitochondria in MSC was analyzed. (G) Primary BM MSC were cocultured with CD45+ cells isolated from Dendra2-mito WT mice with/without AMPK inhibitor, P2RX7 receptor inhibitor and Rejuvesol for 16 hours, and the transfer of mitochondria in MSC was analyzed. (H-I) Schematic illustration of lethally irradiated congenic WT CD45.1+ mice transplanted with WT or Cx43Δ/Δ Dendra2-mito HSPC and treated with vehicle control or AMPK inhibitor (BML-275, 10 mg/kg, interperitoneally) on days 5, 6, and 7 posttransplantation, and analyzed 2.5 hours after the last dose (H). Transfer of Dendra2+ mitochondria from donor HSPC to BM Lin−/CD45−/PDGFRα+/Sca-1− cells in WT and H-Cx43Δ/Δ chimeric mice treated with vehicle control or AMPK inhibitor (I). n = 8 mice per group, 2 independent experiments. All in vitro data are the average of 4 to 6 independent experiments. Data represented as mean ± SEM and statistical significance was assessed using 1-way ANOVA. *P < .05, **P < .01 and ***P < .001.
Mitochondria transfer from healthy donor-derived HSPC boosts BM stroma regeneration and hematopoietic engraftment after irradiation
To understand the significance of mitochondrial transfer from HSPC to the stromal ME in terms of hematopoiesis, we tested whether mitochondrial uptake by BM stromal cells improves stromal cell growth and proliferation in vivo. We found that Sca-1− MSC that received donor-derived mitochondria were able to proliferate more as compared with MSC which did not receive donor-derived mitochondria (Figure 5A). Interestingly, the frequency of proliferative MSC containing donor-derived mitochondria was higher at early time points as compared with later time points after TBI. Indeed, BM Sca-1− MSC from chimeras of Cx43Δ/Δ donor BM HSPC had less ability to proliferate after mitochondria transfer as compared with WT chimeric mice (Figure 5A), suggesting that the hematopoietic Cx43-dependent transfer of mitochondria regulates BM MSC proliferation after irradiation. Isolation of mitochondria from WT and Cx43Δ/Δ Dendra2-mito HSPC followed by its coculture with WT MSC further suggest that uptake of functional mitochondria by MSC induces a higher proliferative state but the effect of Cx43 is dependent on cell-mediated transfer and not on intrinsic properties of the mitochondria derived from WT or Cx43-deficient HSPC (Figure 5B; supplemental Figure 7A-B). To link mitochondria uptake–induced recovery of irradiated MSC, we cocultured irradiated MSC with mitochondria isolated from WT and Cx43Δ/Δ Dendra2-mito HSPC and found that the MSC population containing extracellular mitochondria was enriched in mesenchymal progenitors as assessed by the colony-forming fibroblast (CFU-F) formation with reduced apoptosis (Figure 5C-D). Next, to evaluate whether mitochondria transfer from HSPC to MSC boost their recovery in vivo, we performed CFU-F and colony-forming osteoblast/progenitor (CFU-Ob) frequency assays from chimeras of WT or Cx43Δ/Δ Dendra2-mito hematopoietic cells into WT recipients and found improved recovery of both CFU-F and CFU-Ob in WT chimeras as compared with H-Cx43Δ/Δ chimeras (Figure 5E-G; supplemental Figure 7C). Hematopoietic recovery after irradiation and BMT may depend on the recovered stromal ME. To test this, we studied WT and H-Cx43Δ/Δ chimeric mice for hematopoietic reconstitution and found that donor WT hematopoietic chimeras recovered better in terms of leukocyte, neutrophil, and platelet counts in peripheral blood as well as BM content of phenotypically identified LK (Lin−/cKit+/Sca-1−), LSK (Lin−/cKit+/Sca-1+), long-term, and short-term HSC and multipotential progenitors types 2 and 3, upstream of the process of myelopoiesis and megakaryopoiesis (Figure 5H-I; supplemental Figure 7D-F). We also found that the elevated mitochondria transfer by AMPK inhibition is associated with a faster regeneration of the stromal ME (CFU-F and CFU-Ob) (Figure 5J-K) as well as the hematopoietic compartment (Figure 5L), further supporting the role of AMPK as a negative regulator of mitochondria transfer and BM regeneration. To define the long-term effect of mitochondria transfer, recovery of hematopoietic and BM ME was accessed in Dendra2-mito WT and H-Cx43Δ/Δ chimeric mice paired with 5-FU myeloablation and AMPK inhibition at 6 months post-TBI. Our findings reveal that AMPK inhibition markedly increases mitochondria transfer, specifically to Sca-1− MSC, and efficiently regenerates BM mesenchymal niche after 5-FU–induced myeloablation, which further promotes hematopoietic reconstitution (supplemental Figure 8A-D). These results imply that mitochondria transfer from healthy hematopoietic cells to the BM stromal ME is important for boosting the recovery of the MSC and, in return, will improve the reconstitution of the hematopoietic system after myeloablative conditioning. Finally, to assure that the healthy hematopoietic cells induce the recovery of the stroma and the reconstitution of the hematopoietic compartments as a follow up process, we used a second boost of WT Lin−/CD45+ cell transplantation in WT and H-Cx43Δ/Δ chimeric mice 7 days after primary transplant. This boost of hematopoietic progenitor cells further accelerates CFU-F recovery and hematopoietic reconstitution of both WT and Cx43Δ/Δ chimeric animals, thus suggesting that the beneficial effect on the recovery of the stroma results from the hematopoietic cells that transfer their mitochondria (supplemental Figure 8E-F). Altogether, our findings demonstrate that healthy hematopoietic cells transplanted after lethal irradiation, transfer part of their mitochondria to the stromal ME to speed BM mesenchymal regeneration and, in turn, increase the hematopoietic reconstitution, and this process is regulated positively by hematopoietic Cx43 and negatively by AMPK activity.

Mitochondria transfer from healthy hematopoietic progenitors boosts stromal cell proliferation and hematopoietic recovery following irradiation and transplantation. (A) Proliferation rates in WT and H-Cx43Δ/Δ chimeric mice that were injected with BrdU for 1 hour before analysis. Proliferation of BM Lin−/CD45−/PDGFRα+/Sca-1− MSC receiving (positive) or not (negative) Dendra2+ mitochondria from donor hematopoiesis was measured at 10, 17, and 28 days posttransplantation. Data are the average of 4 mice per group. (B) Dendra2+ mitochondria isolated from WT and Cx43Δ/Δ Dendra2-mito Lin-negative cells were cocultured over irradiated (7.5 Gy) WT stroma for 24 and 48 hours. Brdu uptake by MSC containing or not containing Dendra2+ mitochondria were analyzed at indicated times of coculture. Data are the average of 3 to 6 independent experiments. Dendra2+ mitochondria isolated from WT HSPC (WT mito), Dendra2+ mitochondria isolated from Cx43Δ/Δ HSPC (Cx43Δ/Δ mito). (C-D) Dendra2+ mitochondria isolated from WT and Cx43Δ/Δ Lin-negative cells were cocultured over irradiated (7.5 Gy) WT stroma. After 24 hours, MSC containing (Dendra2-mito+) or not (Dendra2-mito−) extracellular Dendra2 mitochondria were FACS sorted and grown for 3 days. CFU-F (C), and apoptosis as measured by Annexin V staining (D) in Dendra2-mito+ and Dendra2-mito− MSC are shown. Data are the average ± SEM of 3 independent experiments. (E-I) Schematic illustration of lethally irradiated congenic WT CD45.1+ mice that were transplanted with WT or Cx43Δ/Δ Dendra2-mito Lin−/CD51− cells and analyzed at days 10, 17, and 28 posttransplantation (E). Representative example (F) and the frequency of BM CFU-F and CFU-Ob in donor, WT, and H-Cx43Δ/Δ chimeric mice at indicated time posttransplantation (G). BM ST-HSC (Lin−/c-Kit+/Sca-1+/CD48−/CD150−) content in WT and H-Cx43Δ/Δ chimeric mice at days 10, 17, and 28 posttransplantation (H). Peripheral blood counts for leukocytes, neutrophils, and platelets in WT and H-Cx43Δ/Δ chimeric mice at indicated times posttransplantation (I). Data are presented as average ± standard deviation from 2 independent experiments using 4 to 8 mice per group. (J-L) Lethally irradiated WT mice transplanted with WT or Cx43Δ/Δ Dendra2-mito HSPC were treated with vehicle control or AMPK inhibitor (BML-275, 10 mg/kg, intraperitoneally) on days 5, 6, and 7 posttransplantation and analyzed. CFU-F (J) and CFU-Ob (K) in vehicle or AMPK inhibitor–pretreated WT and H-Cx43Δ/Δ chimeric mice on day 7 posttransplantation. Peripheral blood platelet and neutrophil counts in WT and H-Cx43Δ/Δ chimeric mice treated with vehicle control (DMSO) or AMPK inhibitor (BML-275) at indicated time points posttransplantation (L). Data are the average of 4 to 6 mice per group, 2 independent experiment. All data represented as mean ± SEM. Statistical significance was assessed using 2-tailed Student t test except in panels A, C-D where 1-way ANOVA was used. *P < .05, **P < .01, ***P < .001. Scale bar, 10 µm.
Mitochondria transfer from healthy hematopoietic progenitors boosts stromal cell proliferation and hematopoietic recovery following irradiation and transplantation. (A) Proliferation rates in WT and H-Cx43Δ/Δ chimeric mice that were injected with BrdU for 1 hour before analysis. Proliferation of BM Lin−/CD45−/PDGFRα+/Sca-1− MSC receiving (positive) or not (negative) Dendra2+ mitochondria from donor hematopoiesis was measured at 10, 17, and 28 days posttransplantation. Data are the average of 4 mice per group. (B) Dendra2+ mitochondria isolated from WT and Cx43Δ/Δ Dendra2-mito Lin-negative cells were cocultured over irradiated (7.5 Gy) WT stroma for 24 and 48 hours. Brdu uptake by MSC containing or not containing Dendra2+ mitochondria were analyzed at indicated times of coculture. Data are the average of 3 to 6 independent experiments. Dendra2+ mitochondria isolated from WT HSPC (WT mito), Dendra2+ mitochondria isolated from Cx43Δ/Δ HSPC (Cx43Δ/Δ mito). (C-D) Dendra2+ mitochondria isolated from WT and Cx43Δ/Δ Lin-negative cells were cocultured over irradiated (7.5 Gy) WT stroma. After 24 hours, MSC containing (Dendra2-mito+) or not (Dendra2-mito−) extracellular Dendra2 mitochondria were FACS sorted and grown for 3 days. CFU-F (C), and apoptosis as measured by Annexin V staining (D) in Dendra2-mito+ and Dendra2-mito− MSC are shown. Data are the average ± SEM of 3 independent experiments. (E-I) Schematic illustration of lethally irradiated congenic WT CD45.1+ mice that were transplanted with WT or Cx43Δ/Δ Dendra2-mito Lin−/CD51− cells and analyzed at days 10, 17, and 28 posttransplantation (E). Representative example (F) and the frequency of BM CFU-F and CFU-Ob in donor, WT, and H-Cx43Δ/Δ chimeric mice at indicated time posttransplantation (G). BM ST-HSC (Lin−/c-Kit+/Sca-1+/CD48−/CD150−) content in WT and H-Cx43Δ/Δ chimeric mice at days 10, 17, and 28 posttransplantation (H). Peripheral blood counts for leukocytes, neutrophils, and platelets in WT and H-Cx43Δ/Δ chimeric mice at indicated times posttransplantation (I). Data are presented as average ± standard deviation from 2 independent experiments using 4 to 8 mice per group. (J-L) Lethally irradiated WT mice transplanted with WT or Cx43Δ/Δ Dendra2-mito HSPC were treated with vehicle control or AMPK inhibitor (BML-275, 10 mg/kg, intraperitoneally) on days 5, 6, and 7 posttransplantation and analyzed. CFU-F (J) and CFU-Ob (K) in vehicle or AMPK inhibitor–pretreated WT and H-Cx43Δ/Δ chimeric mice on day 7 posttransplantation. Peripheral blood platelet and neutrophil counts in WT and H-Cx43Δ/Δ chimeric mice treated with vehicle control (DMSO) or AMPK inhibitor (BML-275) at indicated time points posttransplantation (L). Data are the average of 4 to 6 mice per group, 2 independent experiment. All data represented as mean ± SEM. Statistical significance was assessed using 2-tailed Student t test except in panels A, C-D where 1-way ANOVA was used. *P < .05, **P < .01, ***P < .001. Scale bar, 10 µm.
Discussion
Our data indicate that the BM ME support is partially lost because of myeloablation damage. Because of such damage to the BM ME, healthy transplanted HSPC not only reconstitute the hematopoietic system, but also need to repair their ME in order to survive and maintain their repopulation potential. We show that HSPC can transfer their mitochondria to the damaged BM ME to boost their recovery by metabolically supporting their function. Interestingly, this mitochondrial transfer to BM CD45−/Lin−/PDGFRα+/Sca-1−, a BM ME population enriched in Cxcl12-expressing adventitial reticular cells and mesenchymal progenitors,39-42 is cell contact dependent via HSPC Cx43 and involves AMPK signaling.
Mitochondrial transfer can rescue aerobic respiration.43 Beneficial effects of mitochondrial transfer from MSC to parenchymal cells have been reported in various normal and malignant cell types, including hematopoietic and leukemic cells.23,26,44-47 We show for the first time that mitochondria transfer also occurs in the opposite direction, meaning from hematopoietic progenitor cells to the MSC population and to a higher extent than the transfer from MSC to the hematopoietic cells. This exchange of mitochondria results in Cx43-dependent scavenging of ROS from donor HSPC17 and the support of the metabolic activity of the recipient MSC and their regenerative functionality, which further contribute to the success of the hematopoietic engraftment. Mitochondrial share between healthy and metabolically impaired cells adds another complexity level to the cross talk between cells and especially it is relevant for the dual regulation of HSPC and their stromal ME. Activation of AMPK may be achieved by lower ATP cellular levels, an expected ATP status in aerobic respiration compromised cells and ATP can repress AMPK activation in lymphohematopoietic cells.48 Our data show that P2RX7–dependent ATP sensing upstream of HSPC AMPK controls mitochondria transfer from HSPC to the stromal population. A higher ATP content is indeed expected in cells with healthy mitochondria, which may sustain other metabolically compromised adjacent cells by supplying them with functional mitochondria. It is possible that the inhibition of AMPK derepresses Cx43-independent mechanisms of mitochondrial transfer. Our data strengthen the view that the seesaw between ATP and AMPK activation in cells may determine the directionality of mitochondria transfer between 2 interacting populations. These data are complemented with the recent finding that mitochondria transfer from MSC to T cells can induce their differentiation into regulatory T cells after transplantation,49 a cell type with ability to modulate allogeneic HSC engraftment.50 These results support our observation regarding the role of mitochondria transfer in achieving a successful HSC engraftment after transplantation.
The mitochondria transfer from healthy, lodged BM HSPC to cell-contact–dependent BM MSC environment after TBI and BMT supply the ME with functional mitochondria that not only induce their proliferation but also may contribute to their differentiation and reduce the inflammatory impact of cytokine responses (reviewed in Reference 51).
Finally, our data imply that mitochondria transfer may have clinical implications which include the administration of an AMPK inhibitor before transplantation or the ex vivo culture with inflammatory-signal deprived, cell-targeted mitochondria.
In summary, we identified a novel role for mitochondria transfer from HSPC to their surrounding mesenchymal ME within the BM that is important and beneficial for transplantation procedures. Our study demonstrates a new regulatory cross talk between HSPC and their stromal ME as well as the role of Cx43 as a positive regulator and AMPK as a negative regulator of hematopoietic-to-mesenchymal mitochondrial transfer.
For original data, please contact the corresponding authors, Jose A. Cancelas (jose.cancelas@uc.edu) and Tsvee Lapidot (tsvee.lapidot@weizmann.ac.il).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Flow Cytometry, Microscopy, and Animal Core Facilities of Cincinnati Children’s Hospital Medical Center and the Weizmann Institute for their excellent support.
This project has been funded by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK124115 (J.A.C.), the American Society of Hematology (A.K.S.), the Israel Science Foundation (ISF 458/17; T.L. and K.G.), the Joint Canada-Israel Health Research Program (ISF 3565/19; T.L. and K.G.), and the Weizmann Helen and Martin Kimmel Stem Cell Institute (T.L.).
Authorship
Contribution: K.G., A.K.S., O.K., M.B., M.J.A., E.K.-M., E.P.-K., A.M.W., H.M., S.L.-Z., T.D., B.B., and M.V.G. performed experiments; K.G., A.K.S., O.K., B.D., T.L., and J.A.C. analyzed data; and K.G., A.K.S., O.K., T.L., and J.A.C. designed research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jose A. Cancelas, Hoxworth Blood Center, 3130 Highland Ave, Cincinnati, OH, 45267; e-mail: jose.cancelas@uc.edu; and Tsvee Lapidot, Department of Immunology, Weizmann Institute of Science, Herzl St 234, PO Box 26, Rehovot 7610001, Israel; e-mail: tsvee.lapidot@weizmann.ac.il.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal