

In this issue of Blood, 1 describe how mitochondrial transfer from hematopoietic stem cells (HSCs) to bone marrow (BM) mesenchymal stem cells (MSCs) helps to regenerate the stroma and its capacity to sustain hematopoietic recovery in irradiated recipient mice transplanted with HSCs.
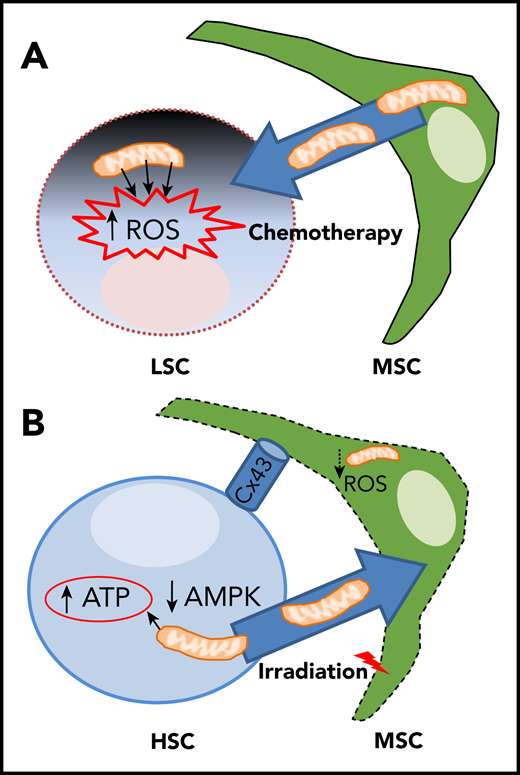
Yin/yang bidirectional exchange of mitochondria between MSC and HSC/LSC during chemotherapy and hematopoietic recovery following TBI. (A) Chemotherapy causes excessive mitochondrial-derived ROS levels in leukemic stem cells (LSC) and triggers mitochondrial donation from MSCs favoring LSC chemoresistance and relapse. (B) TBI reduces mitochondrial function and ROS levels in BM-MSC, but high ATP levels and low AMPK activity in transplanted HSCs promote Cx43- and cell-contact-dependent mitochondrial transfer from HSCs/progenitors to MSCs, favoring both stromal and hematopoietic regeneration.
Yin/yang bidirectional exchange of mitochondria between MSC and HSC/LSC during chemotherapy and hematopoietic recovery following TBI. (A) Chemotherapy causes excessive mitochondrial-derived ROS levels in leukemic stem cells (LSC) and triggers mitochondrial donation from MSCs favoring LSC chemoresistance and relapse. (B) TBI reduces mitochondrial function and ROS levels in BM-MSC, but high ATP levels and low AMPK activity in transplanted HSCs promote Cx43- and cell-contact-dependent mitochondrial transfer from HSCs/progenitors to MSCs, favoring both stromal and hematopoietic regeneration.
Conditioning regimens for clinical HSC transplantation (HSCT) include total body irradiation (TBI) to replace the host’s hematopoietic system with donor-derived cells. Even a low dose of ionizing radiation (frequently used in medical imaging) can increase reactive oxygen species (ROS) and activate p38MAPK in HSCs, compromising their self-renewal capacity.2 In contrast, HSC niche-forming MSCs are much more radioresistant, but cumulative evidence indicates that they do not escape the damage caused by TBI.3 However, the mechanisms mediating HSC niche regeneration are largely unknown. Therefore, they represent an area of intense investigation because understanding these mechanisms would enable optimization of current HSCT protocols.
In a previous study, the authors demonstrated that HSCs/progenitors can be protected from myeloablation-induced oxidative damage through ROS transfer and scavenging by MSCs via the gap junction protein connexin-43 (Cx43).4 In the current study, they show that Cx43 in HSCs/progenitors in direct contact with MSCs allows a previously unrecognized mechanism to foster niche and hematopoietic recovery following TBI and HSCT: mitochondrial transfer from HSCs to BM-MSCs.
First, the authors demonstrate that TBI causes loss of mitochondrial membrane potential and toxic levels of mitochondrial ROS in BM-MSCs, which are progressively reduced in numbers. To visualize and measure mitochondria, they use Dendra2-mitochondria transgenic mice, whereby Dendra2 fluorescent protein is fused with the targeting signal of subunit 8a of mitochondrial cytochrome oxidase. These experiments confirm mitochondrial reduction and fragmentation in MSCs following TBI.1
Through the generation of chimeric mice, the authors demonstrate a bidirectional mitochondrial exchange between donor hematopoietic cells and a subpopulation of Sca1− MSCs. Mitochondria preferentially travel from the donor hematopoietic cells to the latter (ie, host MSCs), with up to 90% MSCs up taking mitochondria from hematopoietic cells over a month. The potential human relevance is suggested by coculture experiments with human CD34+ HSPCs, which transfer mitochondria to mouse stromal cells, albeit at lower frequency (20%), possibly because of the shorter time or to the absence of in vivo signals triggering the mitochondrial exchange.1
Transferred mitochondria are morphologically and functionally integrated in recipient MSCs, where they increase the mitochondrial membrane potential and ROS production. Mitochondrial exchange requires cell–cell contact and the gap junction protein Cx43 (which seems to be needed for metabolic coupling between HSCs/progenitors and Sca1− MSCs, rather than for mitochondrial passage). Because mitochondrial transfer increases upon irradiation, the authors wished to determine the triggers and investigate the role of a key metabolite (adenosine triphosphate [ATP]), its sensor AMP-activated protein kinase (AMPK) and its receptor P2RX7. The experiments indicate that high ATP levels activate P2RX7 and inhibit AMPK to trigger mitochondrial donation from HSCs/progenitors. Consequently, AMPK inhibition increases mitochondrial transfer. Importantly, this is associated with increased regeneration of the BM stroma after irradiation or myeloablation induced by 5-fluorouracil, improving hematopoietic recovery.
The elegant study by Golan et al opens up numerous questions in this emerging field of intercellular communication. Interestingly, other HSC niche cells (such as endothelial cells) also appear to uptake mitochondria from HSCs, albeit with different kinetics and independently of Cx43 (which mainly favors a tight contact and possibly metabolic coupling between BM-MSCs and HSCs). Because mitochondrial exchange can take place through tunneling nanotubes or exosomes,5,6 future studies will determine the contribution of different niche cells to protection from chemotherapy or irradiation (eg, by buffering excessive ROS levels) and subsequent hematopoietic recovery.
Notably, only BM-MSCs (and not other stromal cells or endothelial cells) show a dramatic mitochondrial loss after TBI, but why and how BM-MSCs adapt their metabolism to compensate for reduced mitochondrial function after TBI is unclear. Increased glycolysis does not appear to provide the energy needed. Therefore, it would be interesting to test whether the damaged mitochondria are used by BM-MSCs for energy production through mitophagy, perhaps explaining the reduced mitochondrial volume in BM-MSCs caused by TBI. Similarly, it seems appealing to investigate whether and how mitophagy or mitochondrial unfolded protein response following mitochondrial stress trigger mitochondrial donation from HSCs.
Another study has shown that ROS-induced phosphatidylinositol 3-kinase activation drives Cx43-dependent mitochondrial transfer from stromal cells to HSCs in response to infection.7 Therefore, it would be interesting to investigate the role of sensors of infection (ie, Toll-like receptors), ROS, and the metabolic coupling between HSCs and MSCs as drivers of directed organelle exchange. Importantly, mitochondrial donation from MSCs does not only regulate HSCs but also T cells, thereby inducing a regulatory T cell program and restricting the inflammatory response (as an additional mechanism of MSC-induced immunomodulation).8 Because regulatory T cells reduce postirradiation BM injury and facilitate HSC engraftment after transplantation,9 collectively these studies highlight the potential relevance of mitochondrial exchange in the setting of clinical HSCT.
It might be important to consider bidirectional mitochondrial exchange in the context of HSCT for leukemia treatment because the opposite mitochondrial migratory route, from BM-MSCs to acute myeloid leukemia (AML) cells, helps protect AML cells from the effect of chemotherapy.5,6,10 However, how AML cells avoid the damage caused by the excessive ROS production from these mitochondria has remained elusive. The reason appears to be a dual protection mechanism by MSCs, enabling increased bioenergetic capacity and antioxidant defense against excessive ROS.10 Therefore, different mitochondrial exchange mechanisms should be probably considered during therapy to (1) increase the eradication of chemoresistant leukemic cells and (2) boost hematopoietic recovery following HSCT (see figure).
Conflict-of-interest disclosure: The author declares no competing financial interests.
