

Key Points
PAR4 activation requires a coordinated rearrangement of ECL3 and Thr153 in the tethered LBS formed by TM3 and TM7.
PAR4 Leu310 (rs2227376) in ECL3 has lower receptor reactivity compared with Pro310 and correlates with reduced VTE risk in GWAS analysis.

Abstract
Protease-activated receptor 4 (PAR4) mediates sustained thrombin signaling in platelets and is required for a stable thrombus. PAR4 is activated by proteolysis of the N terminus to expose a tethered ligand. The structural basis for PAR4 activation and the location of its ligand binding site (LBS) are unknown. Using hydrogen/deuterium exchange (H/D exchange), computational modeling, and signaling studies, we determined the molecular mechanism for tethered ligand–mediated PAR4 activation. H/D exchange identified that the LBS is composed of transmembrane 3 (TM3) domain and TM7. Unbiased computational modeling further predicted an interaction between Gly48 from the tethered ligand and Thr153 from the LBS. Mutating Thr153 significantly decreased PAR4 signaling. H/D exchange and modeling also showed that extracellular loop 3 (ECL3) serves as a gatekeeper for the interaction between the tethered ligand and LBS. A naturally occurring sequence variant (P310L, rs2227376) and 2 experimental mutations (S311A and P312L) determined that the rigidity conferred by prolines in ECL3 are essential for PAR4 activation. Finally, we examined the role of the polymorphism at position 310 in venous thromboembolism (VTE) using the International Network Against Venous Thrombosis (INVENT) consortium multi-ancestry genome-wide association study (GWAS) meta-analysis. Individuals with the PAR4 Leu310 allele had a 15% reduction in relative risk for VTE (odds ratio, 0.85; 95% confidence interval, 0.77-0.94) compared with the Pro310 allele. These data are consistent with our H/D exchange, molecular modeling, and signaling studies. In conclusion, we have uncovered the structural basis for PAR4 activation and identified a previously unrecognized role for PAR4 in VTE.
Introduction
Protease initiated signaling events are essential for many physiological processes such as platelet aggregation, inflammatory signaling, and regulation of endothelial barrier function.1-3 Protease-activated receptors (PARs) are primary mediators that transmit signal transduction from the protease to the intracellular signaling machinery. The 4 PAR members, PAR1, -2, -3, and -4, are expressed in many cell types and are activated by a growing panel of proteases.4-8 PARs are activated by enzymatic cleavage of the N terminus to expose a tethered ligand.4,9,10 Compared with a soluble ligand, the attached tethered ligand reduces the increase in entropy upon dissociation, resulting in an entropy-driven higher affinity for PAR activation. This activation mechanism creates unique challenges in targeting PARs therapeutically. Furthermore, because the ligand is attached to the receptor, there are likely substantial conformational changes that occur during PAR activation. These structural rearrangements and the precise mechanism of this unique protein-tethered ligand interaction are unknown.
PAR4 has primarily been studied on platelets where it is co-expressed with PAR1. Together they mediate the full spectrum of thrombin signaling.11 PAR1 is efficiently cleaved by thrombin and mediates rapid transient signaling. In contrast, PAR4 leads to prolonged signaling and sustained platelet activation. Recently, these differences in signaling kinetics have been exploited therapeutically. Blocking PAR4 spares the rapid response mediated by PAR1 thereby limiting the bleeding complications.12 In addition, polymorphisms in the PAR4 gene (f2rl3) lead to sequence variants that impact its signaling and response to some antagonists.13 PAR4 therapies have focused on coronary artery disease. However, there is growing evidence that platelets have a key initiating role in venous thromboembolism (VTE). Despite the key role of PAR4 in platelet biology, its role in VTE and potential as a therapeutic target are unknown.
Despite numerous studies of PAR function, the understanding of the structural basis for the tethered ligand has been limited to nuclear magnetic resonance imaging of the isolated N-terminal exodomain of PAR1 in solution and the crystal structure of PAR1 or PAR2 bound to antagonist.14-16 The major limitation of the crystallization studies is the need for significant modifications to stabilize the receptor in a single conformation. This precludes experiments to identify the endogenous tethered ligand binding site (LBS) and the conformational dynamics upon activation. Amide hydrogen/deuterium exchange (H/D exchange) mass spectrometry (MS) is a structural approach that monitors the deuterium uptake at amide NH groups along the backbone of the protein by using MS.17,18 The power of H/D exchange comes from comparing the deuterium uptake between 2 states to uncover the dynamics of ligand binding or receptor activation.17-23 The distinct advantage of H/D exchange for G protein-coupled receptors (GPCRs) is that it overcomes the need for a stabilized receptor and allows us to examine the conformational dynamics of the tethered ligand mechanism.
Here, we describe the first biophysical studies of the PAR tethered ligand activation mechanism. We applied H/D exchange and computational modeling to uncover the endogenous LBS and a previously unknown function of extracellular loop 3 (ECL3). We verified the role of these regions with cell-based signaling assays. Taken together, we discovered a novel role of the ECL3 in PAR4 activation and demonstrated that the rigidity of this loop is critical to PAR4 activation. Finally, a polymorphism (rs2227376) that changes Pro to Leu in ECL3 significantly lowers PAR4 reactivity and is associated with a 15% reduction of relative risk of VTE.
Materials and methods
Protein expression and purification
Full-length human PAR4 containing an N-terminal FLAG tag was expressed and purified from Sf9 cells as described previously.24 The detailed purification protocols for both PAR4 and G-protein complex are described in the supplemental Data, available on the Blood Web site. The activity of purified PAR4 was verified as described in the supplemental Data.
Amide H/D exchange
Amide H/D exchange was performed and analyzed as described previously.20,25 A detailed H/D exchange procedure, software list, and analysis pipeline are described in the supplemental Data. The H/D exchange was normalized to the maximal deuterium uptake at 10 minutes and the PAR4 homology model is color-coded as indicated in the legends for Figures 2 and 3. Gap regions are colored in gray. The difference map of the deuterium uptake level between nonactivated and thrombin activated contains only the peptides found in both states.
Computational modeling
The computational model of PAR4 in its thrombin-activated state was generated by using the crystal structure of rhodopsin at 2.2 Å resolution (Protein Data Bank entry 1U19) as described for green cone opsin.20 The 2 sequences were aligned using the EMBOSS Needle Pairwise Sequence Alignment tool, and the initial model was generated using the MEDELLER server.26,27 This initial model was imported into the ROSETTA protein structure prediction suite, and the loop building protocol was applied to determine the model with the lowest free energy. Additional details are provided in the supplemental Data.
Generation of PAR4-expressing cell lines
PAR4-wild-type (WT) or PAR4 mutants (PAR4-T153A, PAR4-T153S, PAR4-P310L, PAR4-S311A, and PAR4-P312L) containing an N-terminal V5 epitope were stably expressed using HEK293 Flp-In T-REx cells according to the manufacturer’s protocol (Invitrogen). The concentration of tetracycline was titrated (25-1000 ng/mL) to yield similar expression levels of each PAR4 mutant on the cell surface (∼175 000 receptors per cell) at 40 hours as determined by quantitative flow cytometry28-30 (described in supplemental Data).
Measurement of intracellular calcium flux
PAR4-expressing HEK293 Flp-In T-REx cells were induced for 40 hours, removed from the plate and washed 3 times with N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)-Tyrode’s buffer (pH 7.4). Cells were incubated with 0.5 μM Fura-2 and 100 nM vorapaxar for 1 hour. The cells were then washed 3 times in HEPES-Tyrode’s buffer with 200 μM CaCl2 and diluted to 2.5 × 105 cells per mL. The intracellular calcium mobilization was recorded using a fluorometer (Photon Technology International, Inc.) for 420 seconds total. The first 50 seconds was set as the background, the response to agonist was measured for 250 seconds, and the maximum and minimum fluorescence was measured for 60 seconds each by adding 0.1% Triton X100 and 8.8 mM EDTA sequentially.28,31,32 Fluorescence values were converted to intracellular free Ca2+ concentration with the equations described by Grynkiewicz et al.31 A more detailed protocol is described in the supplemental Data.
Results
Analysis of PAR4 activation with H/D exchange
Full-length human PAR4 was expressed and purified in Sf9 cells as described previously.24 The protein was purified with affinity chromatography followed by size exclusion chromatography (Figure 1A). The purity of the protein was confirmed by both Coomassie staining and western blot analysis (Figure 1B). The dithiothreitol (DTT) in the purification procedure has the potential to affect the re-oxidation of the disulfide bond that connects ECL1 to ECL2 and influence the results of the H/D exchange. Therefore, we used a G-protein activation assay to confirm that the purified protein retains activity, which would indicate that the detergent and DTT present in the purification did not have an impact on protein folding (Figure 1C). The tandem mass spectrometry spectra of PAR4 digested by pepsin were analyzed by MassMatrix software. The peptide mapping studies had 91.2% (344 of 377) amino acid sequence coverage of the deuterated PAR4 (Figure 2A). The deuterium uptake of PAR4 ranged from 0% to 66%, which agrees with other GPCRs.20,33,34

Human PAR4 expression and purification. Full-length human PAR4 was expressed and purified from Sf9 cells. The protein was purified by FLAG antibody affinity chromatography followed by size exclusion chromatography. (A) A size exclusion chromatogram was obtained from purifications. Human PAR4 with a C-terminal 1D4 tag eluted in the main peak from fractions 14 to 20 (0.5 mL per fraction). The other small peaks contain tobacco etch virus for cutting the b562 RIL (BRIL) sequence from PAR4, the BRIL sequence, and the FLAG peptide for affinity chromatography. (B) Using both Coomassie staining (left) and immunoblotting with 1D4 antibody (right), the purity of the purified human PAR4 was confirmed. Purified PAR4 is a 37-kDa band on reduced sodium dodecyl sulfate-polyacrylamide gel electrophoresis with an extra glycosylated PAR4 band. The thrombin cleavage removed 2.9 kDa from the protein, and the activated PAR4 is at 34 kDa. (C) A G-protein assay measured the relative fluorescence change of tryptophan in the PAR4 and G-protein complex to confirm that the purified PAR4 is functional.
Human PAR4 expression and purification. Full-length human PAR4 was expressed and purified from Sf9 cells. The protein was purified by FLAG antibody affinity chromatography followed by size exclusion chromatography. (A) A size exclusion chromatogram was obtained from purifications. Human PAR4 with a C-terminal 1D4 tag eluted in the main peak from fractions 14 to 20 (0.5 mL per fraction). The other small peaks contain tobacco etch virus for cutting the b562 RIL (BRIL) sequence from PAR4, the BRIL sequence, and the FLAG peptide for affinity chromatography. (B) Using both Coomassie staining (left) and immunoblotting with 1D4 antibody (right), the purity of the purified human PAR4 was confirmed. Purified PAR4 is a 37-kDa band on reduced sodium dodecyl sulfate-polyacrylamide gel electrophoresis with an extra glycosylated PAR4 band. The thrombin cleavage removed 2.9 kDa from the protein, and the activated PAR4 is at 34 kDa. (C) A G-protein assay measured the relative fluorescence change of tryptophan in the PAR4 and G-protein complex to confirm that the purified PAR4 is functional.
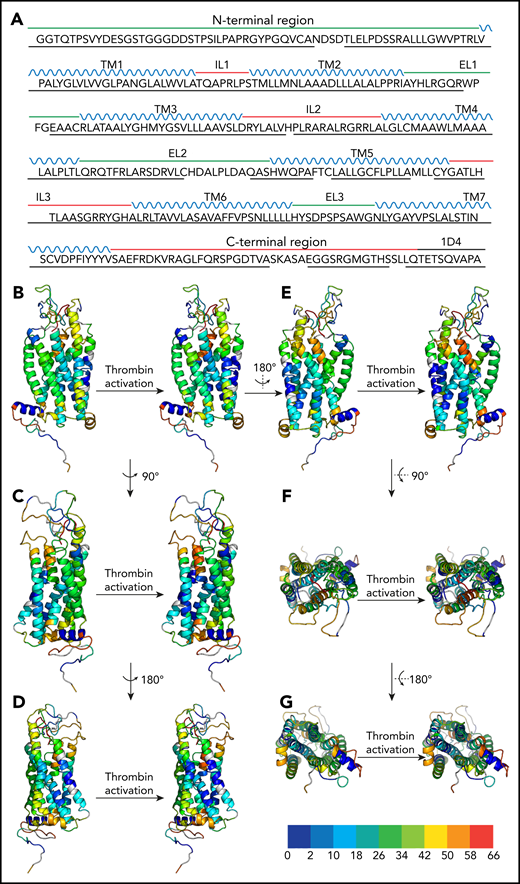
Peptide coverage of the human PAR4 construct by MS and H/D exchange map of the apo and thrombin-activated states of human PAR4. (A) Green line, the extracellular side of the PAR4 sequence; red line, the intracellular domain of PAR4; blue, the transmembrane domains; black, the C-terminal 1D4 tag. The black underline indicates the peptide coverage in H2O, D2O apo state, and D2O thrombin-activated state. Only the peptides that are shown in all 3 states and that have a significant score with a P value <.05 were selected and further analyzed. (B-E) Side view of PAR4 in apo and thrombin-activated states. (F-G) Top and bottom views of PAR4 in apo and thrombin-activated states, respectively. In all panels, the apo state of PAR4 is on the left and the thrombin-activated state is on the right. The PAR4 homology model is color-coded on the basis of the H/D exchange level: dark blue for 0% to <2%, light blue for 2% to <10%, cyan for 10% to <18%, cadet blue for 18% to <26%, green for 26% to <34%, light green for 34% to <42%, yellow for 42% to <50%, orange for 5% to <58%, and red for 58% to 66%. Undefined regions are gray.
Peptide coverage of the human PAR4 construct by MS and H/D exchange map of the apo and thrombin-activated states of human PAR4. (A) Green line, the extracellular side of the PAR4 sequence; red line, the intracellular domain of PAR4; blue, the transmembrane domains; black, the C-terminal 1D4 tag. The black underline indicates the peptide coverage in H2O, D2O apo state, and D2O thrombin-activated state. Only the peptides that are shown in all 3 states and that have a significant score with a P value <.05 were selected and further analyzed. (B-E) Side view of PAR4 in apo and thrombin-activated states. (F-G) Top and bottom views of PAR4 in apo and thrombin-activated states, respectively. In all panels, the apo state of PAR4 is on the left and the thrombin-activated state is on the right. The PAR4 homology model is color-coded on the basis of the H/D exchange level: dark blue for 0% to <2%, light blue for 2% to <10%, cyan for 10% to <18%, cadet blue for 18% to <26%, green for 26% to <34%, light green for 34% to <42%, yellow for 42% to <50%, orange for 5% to <58%, and red for 58% to 66%. Undefined regions are gray.
The overall analysis identified conserved regions essential for GPCR signaling. As expected, the N- and C-termini had high deuterium uptake, indicating water accessibility; and the 7 transmembrane (TM) domains of PAR4 in both uncleaved (apo) state and activated state had limited deuterium uptake because they were being buried in detergent micelles (Figure 2B-G). Notably, TM4 and TM5 had less deuterium uptake compared with the other TM helices, suggesting that the dimerization interface of PAR4 was formed by TM4 and TM5 (supplemental Figure 1). This is in agreement with our previous biochemical studies, which also mapped the dimer interface to TM4, specifically to residues L1924.41, L1944.43, M1984.47, and L2024.51 (Ballesteros and Weinstein nomenclature).29,30 Furthermore, the deuterium uptake at the dimer interface (M2034.52-L2134.62) decreased from 6.4% to 3.4% upon cleavage by thrombin, suggesting that the PAR4 dimer packed more tightly upon activation (supplemental Figure 1; supplemental Table 1). Intracellular loop 2 (ICL2) is the docking site for the heterotrimeric G-protein or arrestin and is essential for the propagation of intracellular signaling.35-41 The H/D exchange of ICL2 (H180PLRA184) increased from 41% to 45% upon activation, which indicates that this loop had more solvent accessibility in the activated state (Figures 2B-G and 3; supplemental Table 1). These data suggest that ICL2 becomes accessible after receptor activation allowing the G-protein or arrestin to dock at its binding site. Helix 8 is a short α-helix between TM7 and the C terminus that may contribute to GPCR localization, receptor internalization, and/or G-protein activation.42 The deuterium uptake in both apo and activated states was low (0.18%), indicating that it remains buried in the detergent (Figures 2B-G and 3; supplemental Table 1). However, the deuteration at the hinge region (amino acids Y342 and VSAEF347) increased from 41% to 50%, showing that helix 8 is flexible in reference to the 7 transmembrane helical bundle (supplemental Table 1).
![H/D exchange difference map of human PAR4 in apo and thrombin-activated states. The percent deuteration of the thrombin-activated state was subtracted from the percent deuteration of the apo state. The PAR4 homology model was color-coded on the basis of the difference in percent deuteration between the 2 states (Δ % deuteration [apo-activated]): dark blue for –9% to <−7%, light blue for −7% to <−5%, cyan for –5% to <−3%, cadet blue for –3% to <−1%, green for –1% to <1%, light green for 1% to <3%, yellow for 3% to <5%, orange for 5% to <7%, and red for 7% to 9+%. Undefined regions are gray. (A-D) Side views of PAR4. (E) Top view and (F) bottom view.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/19/10.1182_blood.2019004634/2/m_bloodbld2019004634f3.png?Expires=1763476536&Signature=1BZV7yrFU7XLEZYcyyzv7x3eiOqYguxrajbHSLNYfvFfV~JkW5tZqg4AS1KJIuKRG~F3A~~tvyZROtoWi0Jhu3hwhUPc1AzNl59D3scFSqufxD69UTACwXfUcxt4X3TAxp~huv-aoM83mk91AJWwmEaxqYumitCVVcRkoGC9xXCjcXfyI2SBfTEw~XSKBTCiCcOgtS3pL4aNC6Cca18xjhtInKVGAdC0Fuba2f-gZ2MnCbSk~bGZxxcRoUC4TMqUo9IjWhWgR5N51VrSfRjeoxdE762XJ~LJtLfzgOgYqlAeCFzJYO0hFcuCPgnQDC-Gzj63CvZsIms0CpauPa79rA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
H/D exchange difference map of human PAR4 in apo and thrombin-activated states. The percent deuteration of the thrombin-activated state was subtracted from the percent deuteration of the apo state. The PAR4 homology model was color-coded on the basis of the difference in percent deuteration between the 2 states (Δ % deuteration [apo-activated]): dark blue for –9% to <−7%, light blue for −7% to <−5%, cyan for –5% to <−3%, cadet blue for –3% to <−1%, green for –1% to <1%, light green for 1% to <3%, yellow for 3% to <5%, orange for 5% to <7%, and red for 7% to 9+%. Undefined regions are gray. (A-D) Side views of PAR4. (E) Top view and (F) bottom view.
H/D exchange difference map of human PAR4 in apo and thrombin-activated states. The percent deuteration of the thrombin-activated state was subtracted from the percent deuteration of the apo state. The PAR4 homology model was color-coded on the basis of the difference in percent deuteration between the 2 states (Δ % deuteration [apo-activated]): dark blue for –9% to <−7%, light blue for −7% to <−5%, cyan for –5% to <−3%, cadet blue for –3% to <−1%, green for –1% to <1%, light green for 1% to <3%, yellow for 3% to <5%, orange for 5% to <7%, and red for 7% to 9+%. Undefined regions are gray. (A-D) Side views of PAR4. (E) Top view and (F) bottom view.
H/D exchange also identified novel regions unique to the tethered ligand mechanism of PAR4. On the basis of the current model of PAR activation, we predicted the N terminus would have a high deuterium uptake in the apo state that is reduced after the tethered ligand binds to a receptor. Surprisingly, in our H/D exchange studies, the PAR4 tethered ligand G48YPGQV had a low deuterium uptake in both apo and thrombin-activated states (Figures 2B-G and 3; supplemental Table 1), suggesting the tethered ligand docks near the extracellular loops before thrombin activation.
Comparing PAR4 in the apo and thrombin-activated states revealed 2 regions with a large change in deuteration upon activation (Figure 3). First, a region formed by amino acids C1493.25-L1563.32 in TM3 and S3297.42-S3337.46 in TM7 had significantly lower deuteration upon thrombin activation, suggesting that this is the LBS in which water accessibility was blocked by the tethered ligand upon proteolytic activation. Second, ECL3 and the adjacent regions of TM6 and TM7 (F2966.48-A3277.40) had increased deuterium uptake after thrombin activation (Figure 3). Given that this loop is located near the identified LBS, ECL3 may act as a gatekeeper to prevent access of the tethered ligand to its binding site before thrombin activation of PAR4.
Computational modeling
To further validate the H/D exchange studies, a human PAR4 homology model in the thrombin-activated state was generated on the basis of the crystal structure of the prototypical Class A GPCR rhodopsin at a resolution of 2.2 Å.20,43 The computational modeling was based on an original unbiased search of 10 000 computational models filtered by energetic allowance and optimized by searching for the energy landscape. The model with the lowest free energy level was selected. In this model, Gly48 in the tethered ligand interacts with Thr153 (Thr3.29) in the LBS identified in the H/D exchange studies (Figure 4). This raised the hypothesis that Thr153 has a critical role in PAR4 activation by forming a direct interaction with Gly48 from the tethered ligand upon thrombin activation.

Structural modeling showing the interaction between the tethered ligand and the LBS. The homology model for PAR4 in the activated state was generated by computational modeling. In this model, the tethered ligand is green, the LBS is red, and Gly48 and Thr153 are cyan. The TM4 and part of ECL2 are hidden on the homology model to provide a better view of the LBS.
Structural modeling showing the interaction between the tethered ligand and the LBS. The homology model for PAR4 in the activated state was generated by computational modeling. In this model, the tethered ligand is green, the LBS is red, and Gly48 and Thr153 are cyan. The TM4 and part of ECL2 are hidden on the homology model to provide a better view of the LBS.
Thr153 is critical for PAR4 ligand binding
H/D exchange and computational modeling suggested that Thr153 is critical for PAR4 activation. To test this in cells, we generated inducible cell lines stably expressing PAR4-WT, PAR4-T153A, or PAR4-T153S. To allow a direct comparison between each PAR4 mutant, the expression level of each construct was tuned by adjusting the tetracycline concentration to achieve ∼175 000 receptors per cell as determined by quantitative flow cytometry (Figure 5A). Because HEK293 cells endogenously express PAR1, vorapaxar (100 nM) was used to block Ca2+ signaling from thrombin-activated PAR1 (supplemental Figure 2A-B); PAR1 inhibition was verified in all subsequent experiments (supplemental Figure 2C-N). We also confirmed that HEK293 cells do not respond to the PAR4-specific activation peptide (AYPGKF-NH2) (supplemental Figure 2A).
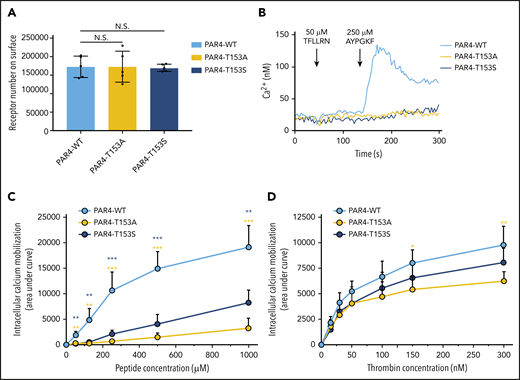
Thr153in the LBS is critical for PAR4 activation. (A) Comparable expression levels of PAR4-WT, PAR4-T153A, and PAR4-T153S on the surface of the HEK293 Flp-In T-REx stable cell lines confirmed by flow cytometry using a V5-fluorescein isothiocyanate (FITC)–conjugated antibody. (B) HEK293 cell expresses PAR1 endogenously, which is inhibited by 100 nM vorapaxar. There is no intracellular calcium mobilization triggered by stimulating HEK293 Flp-In T-REx cells that express PAR4-WT, PAR4-T153A, or PAR4-T153S with 50 μM PAR1-AP (TFLLRN). Stimulating PAR4-WT with 250 μM of PAR4-AP initiated calcium flux, but PAR4-T153A and PAR4-T153S had no response to this dose of PAR4-AP. (C) PAR4-WT, PAR4-T153A, and PAR4-T153S were challenged with 5 different doses of PAR4-AP, and calcium mobilization responses to PAR4-AP stimulation were measured (n = 5). Data are presented as the area under curve (AUC) after stimulation of the Fura2-loaded HEK293 Flp-In T-REx stable cell lines. (D) PAR4-WT, PAR4-T153A, and PAR4-T153S were challenged with 6 different doses of α-thrombin, and calcium mobilization responses to α-thrombin stimulation were measured (n = 5). Data are presented as the AUC after stimulation of the Fura2-loaded HEK293 Flp-In T-REx stable cell lines. Data are mean ± standard deviation (SD) from 5 independent experiments at each concentration. N.S., not significant, unpaired Student t test. *P < .05; **P < .01; ***P < .001. The yellow asterisks indicate the comparison of PAR4-T153A to PAR4-WT and the blue asterisks indicate the comparison of PAR4-T153S to PAR4-WT.
Thr153in the LBS is critical for PAR4 activation. (A) Comparable expression levels of PAR4-WT, PAR4-T153A, and PAR4-T153S on the surface of the HEK293 Flp-In T-REx stable cell lines confirmed by flow cytometry using a V5-fluorescein isothiocyanate (FITC)–conjugated antibody. (B) HEK293 cell expresses PAR1 endogenously, which is inhibited by 100 nM vorapaxar. There is no intracellular calcium mobilization triggered by stimulating HEK293 Flp-In T-REx cells that express PAR4-WT, PAR4-T153A, or PAR4-T153S with 50 μM PAR1-AP (TFLLRN). Stimulating PAR4-WT with 250 μM of PAR4-AP initiated calcium flux, but PAR4-T153A and PAR4-T153S had no response to this dose of PAR4-AP. (C) PAR4-WT, PAR4-T153A, and PAR4-T153S were challenged with 5 different doses of PAR4-AP, and calcium mobilization responses to PAR4-AP stimulation were measured (n = 5). Data are presented as the area under curve (AUC) after stimulation of the Fura2-loaded HEK293 Flp-In T-REx stable cell lines. (D) PAR4-WT, PAR4-T153A, and PAR4-T153S were challenged with 6 different doses of α-thrombin, and calcium mobilization responses to α-thrombin stimulation were measured (n = 5). Data are presented as the AUC after stimulation of the Fura2-loaded HEK293 Flp-In T-REx stable cell lines. Data are mean ± standard deviation (SD) from 5 independent experiments at each concentration. N.S., not significant, unpaired Student t test. *P < .05; **P < .01; ***P < .001. The yellow asterisks indicate the comparison of PAR4-T153A to PAR4-WT and the blue asterisks indicate the comparison of PAR4-T153S to PAR4-WT.
Stimulation of PAR4-WT–expressing cells with 250 μM of the PAR4 activation peptide (AYPGKF-NH2) led to a robust Ca2+ signal that was abolished by mutating Thr153 to Ala or Ser; a representative tracing is shown in Figure 5B. Furthermore, both PAR4-T153A and PAR4-T153S had a significant reduction of total Ca2+ flux as determined by area under the curve at all AYPGKF-NH2 concentrations tested (30-1000 μM) (Figure 5C) and peak Ca2+ (supplemental Figure 3A). We next examined the response to the endogenous tethered ligand after activation by thrombin. There was a significant reduction in Ca2+ mobilization in PAR4-T153A–expressing cells when stimulated with 150 and 300 nM thrombin (Figure 5D; supplemental Figure 3B). PAR4-T153S had an intermediate response (Figure 5D; supplemental Figure 3B). These data show that mutating Thr153 reduced the response to both AYPGKF-NH2 and thrombin but was less dramatic for thrombin. Taken together, Ca2+ signaling assays confirmed that Thr153 within the LBS identified by H/D exchange and predicted in the computational modeling is critical for PAR4 activation.
P310 on ECL3 of PAR4 is important for receptor activation
ECL3 had increased deuteration after thrombin activation, which indicates that it becomes exposed to solvent (Figure 3). On the basis of the location of this loop relative to the PAR4 LBS, we hypothesized that it acts as a gatekeeper that swings out after activation by thrombin, which allows the tethered ligand to access the binding pocket. The computational modeling of thrombin-activated PAR4 demonstrated that ECL3 adopts in an “out” position further supporting the gatekeeper hypothesis (supplemental Movie 1). Eight of the 10 residues in ECL3 are identical among human, mouse, and rat PAR4, which suggests a conserved function between species (supplemental Figure 4). Notably, there are 2 proline residues located in ECL3. Prolines located in loops often have a critical role in maintaining the rigidity of these regions.44 Therefore, we postulated that Pro310 and Pro312 are important for maintaining the loop rigidity for PAR4 activation.
We have previously shown that naturally occurring variants in human PAR4 alter receptor reactivity and affect platelet function.13 Coincidently, there is an uncharacterized polymorphism of PAR4 (rs2227376) that changes proline to leucine at position 310 (P310L) in ECL3. We postulated that substituting a leucine for proline in ECL3 would lead to increased flexibility and affect the structural integrity of ECL3, which would have an impact on PAR4 activation and signaling. To test this experimentally, we generated a stable cell line expressing PAR4-P310L (Figure 6A). Mutating Pro310 to Leu abolished total Ca2+ signaling in response to the AYPGKF-NH2 peptide as determined by AUC (Figure 6B-C) and peak Ca2+ (supplemental Figure 5A). The response to thrombin was also significantly reduced (Figure 6D; supplemental Figure 5B). Next, we determined whether other residues in ECL3 contribute to PAR4 function. Pro310 is found within a conserved sequence (P310SP) (supplemental Figure 4). We generated PAR4-S311A, which is predicted to have no impact on the rigidity of ECL3, and PAR4-P312L, which is predicted to increase the flexibility of ECL3. Mutating Ser311 to Ala did not impact Ca2+ signaling in response to AYPGKF-NH2 or thrombin (Figure 6B-D; supplemental Figure 5). Interestingly, mutated Pro312 had an intermediate response to AYPGKF-NH2 compared with PAR4-WT and PAR4-P310L (Figure 6B-C; supplemental Figure 5A). PAR4-P312L seemed to be hyperreactive in response to thrombin, but it did not reach statistical significance (Figure 6D; supplemental Figure 5B). Taken together, these data indicate that the intact rigidity of ECL3 is essential for PAR4 activation.
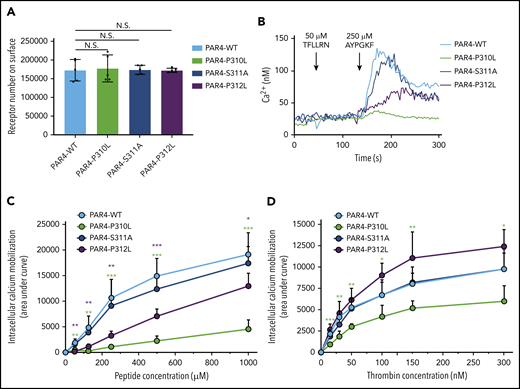
The intact rigidity of ECL3 is essential for the interaction between the tethered ligand and the binding site. (A) Comparable expression levels of PAR4-WT, PAR4-P310L, PAR4-S311A, and PAR4-P312L on the surface of the HEK293 Flp-In T-REx stable cell lines were confirmed by flow cytometry using a V5-FITC–conjugated antibody. (B) Endogenous PAR1 in HEK293 cells is inhibited by 100 nM vorapaxar. No intracellular calcium mobilization was triggered by stimulating HEK293 Flp-In T-REx cells that express PAR4-WT, PAR4-P310L, PAR4-S311A, and PAR4-P312L with 50 μM PAR1-AP. Stimulating PAR4-WT with 250 μM of PAR4-AP initiated calcium flux, and PAR4-P310L had a small but unmistakable response to the same dose of PAR4-AP. At this dose of PAR4-AP, PAR4-S311A cells showed a receptor reactivity similar to that of PAR4-WT, and PAR4-P312L had a hyporesponse to PAR4-AP stimulation. (C) PAR4-WT, PAR4-P310L, PAR4-S311A, and PAR4-P312L were challenged with 5 different doses of PAR4-AP, and calcium mobilization responses to PAR4-AP stimulation were measured (n = 5). Data are presented as the AUC after stimulation of the Fura2-loaded HEK293 Flp-In T-REx stable cell lines. (D) PAR4-WT, PAR4-P310L, PAR4-S311A, and PAR4-P312L were challenged with 6 different doses of α-thrombin, and calcium mobilization responses to thrombin stimulation were measured (n = 5). Data are presented as the AUC after stimulation of the Fura2-loaded HEK293 Flp-In T-REx stable cell lines. Data are mean ± SD from 5 independent experiments at each concentration. Unpaired Student t test; *P < .05; **P < .01; ***P < .001.
The intact rigidity of ECL3 is essential for the interaction between the tethered ligand and the binding site. (A) Comparable expression levels of PAR4-WT, PAR4-P310L, PAR4-S311A, and PAR4-P312L on the surface of the HEK293 Flp-In T-REx stable cell lines were confirmed by flow cytometry using a V5-FITC–conjugated antibody. (B) Endogenous PAR1 in HEK293 cells is inhibited by 100 nM vorapaxar. No intracellular calcium mobilization was triggered by stimulating HEK293 Flp-In T-REx cells that express PAR4-WT, PAR4-P310L, PAR4-S311A, and PAR4-P312L with 50 μM PAR1-AP. Stimulating PAR4-WT with 250 μM of PAR4-AP initiated calcium flux, and PAR4-P310L had a small but unmistakable response to the same dose of PAR4-AP. At this dose of PAR4-AP, PAR4-S311A cells showed a receptor reactivity similar to that of PAR4-WT, and PAR4-P312L had a hyporesponse to PAR4-AP stimulation. (C) PAR4-WT, PAR4-P310L, PAR4-S311A, and PAR4-P312L were challenged with 5 different doses of PAR4-AP, and calcium mobilization responses to PAR4-AP stimulation were measured (n = 5). Data are presented as the AUC after stimulation of the Fura2-loaded HEK293 Flp-In T-REx stable cell lines. (D) PAR4-WT, PAR4-P310L, PAR4-S311A, and PAR4-P312L were challenged with 6 different doses of α-thrombin, and calcium mobilization responses to thrombin stimulation were measured (n = 5). Data are presented as the AUC after stimulation of the Fura2-loaded HEK293 Flp-In T-REx stable cell lines. Data are mean ± SD from 5 independent experiments at each concentration. Unpaired Student t test; *P < .05; **P < .01; ***P < .001.
PAR4-310L allele correlates with a reduced risk of VTE
PAR4 single nucleotide polymorphisms (SNPs) are known to have an impact on platelet function.13 Recently, it was recognized that platelets have a role in the initiation of venous thrombosis, so we examined the correlation of rs2227376 (PAR4-310P/L) and rs773902 (PAR4-120A/T) with VTE. The International Network Against Venous Thrombosis (INVENT) Consortium is a multi-ancestry genome-wide association study meta-analysis with 30 234 VTE cases and 172 122 controls and has been used to identify novel genetic markers for VTE.45 The minor allele frequency for rs2227376 was 0.015 for individuals with European ancestry, and this polymorphism was not present in participants with African ancestry. The PAR4 allele encoding for Leu at 310 had a 15% reduction in relative risk for VTE (odds ratio, 0.85; 95% confidence interval, 0.77-0.94) compared with the Pro for each Leu allele. Protection from VTE is consistent with the Leu having decreased PAR4 reactivity. The minor allele frequency for rs773902 was 0.19 in those with European ancestry and 0.45 in those with African ancestry, which is consistent with previous reports. However, rs773902 was not associated with VTE in the INVENT data: odds ratio, 1.02; 95% confidence interval, 0.99-1.05).
Discussion
PARs are important mediators that facilitate communication between proteases and the intracellular signaling machinery. Since PARs were discovered, their roles in a wide range of processes have been intensely studied and are well characterized.4-9,46-49 However, the biophysical and structural basis for the activation mechanism of this receptor family is still limited. In addition, the location of the tethered LBS of PAR is unclear. Here, we applied amide H/D exchange MS combined with computational modeling and cell signaling to uncover the location of the endogenous LBS and the structural basis for the tethered ligand–mediated activation mechanism for PAR4.
We and others have previously shown that PAR4 forms homodimers and heterodimers with PAR1, PAR3, and P2Y12.29,30,50-55 These physical interactions have an impact on receptor activation, signaling, and response to antagonists. We observed less deuterium uptake in TM4 and TM5 compared with the other helices. The tight packing arrangement of this region suggests a homodimer interface (supplemental Figure 1). Consistent with these data, we previously mapped the PAR4 homodimer interface to TM4 using biochemical and signaling studies.30 The heterodimer interface of PAR4 with PAR1 and P2Y12 have also been mapped to TM4.29,50 The agreement of our H/D exchange data with published studies of PARs and other GPCRs gives us confidence that our recombinant protein accurately represents PAR4 expressed on cells.51,56-59
The precise nature of the interactions between the tethered ligand and the receptor has remained elusive. Initial studies with PAR1 and PAR2 revealed a role for ECL2.10,60,61 Nanevicz et al10 showed a complementary relationship between ECL2 and the tethered ligand in elegant studies using a panel of human-Xenopus PAR1 chimeric receptors.10,60 Mutagenesis studies have also identified key residues in ECL2 for PAR2.61 Our H/D exchange difference map identified a binding pocket formed by TM3 and TM7 on the extracellular side of the receptor that had significantly less deuterium uptake in the thrombin-activated PAR4. This suggests that the tethered ligand blocked solvent accessibility to this region (Figure 3). In addition, ECL3 had increased deuterium uptake after activation, which indicated an increase in solvent accessibility of this loop. Computational modeling confirmed the H/D exchange showing that Gly48 of the tethered ligand interacts with Thr153 in the binding pocket and that ECL3 is in an open conformation (Figure 4). These data support a unique mechanism for PAR4 activation by the tethered ligand that involves a deep binding pocket within the TM helices and ECL3, which has not been identified for PAR1 and PAR2. PAR4 has signaling characteristics distinct from those of PAR1 and PAR2. PAR4 mediates prolonged signaling in platelets and other cells.11,62,63 In contrast, PAR1 and PAR2 mediate transient signaling.61-63 We propose that the deep binding pocket we identified may contribute to the sustained signaling of PAR4, whereas the transient signaling of PAR1 and PAR2 may be mediated by transient interactions with ECL2 on the surface of the receptor. Our H/D exchange and modeling did not reveal a role for ECL2. However, a recent study by Thibeault et al64 showed that His229 and Asp230 in ECL2 are critical for PAR4 activation. This discrepancy may be a result of interactions between the exodomain and extracellular loops before activation that prevent H/D exchange. Traditionally, the N terminus of PARs is thought to be unstructured and highly flexible. Therefore, we hypothesized that it would have high deuterium uptake in the apo state that decreased upon activation by the protease. Unexpectedly, the PAR4 exodomain including the tethered ligand (48GYPGQV) had low deuterium uptake in both states, suggesting that the N terminus of PAR4 docks near the extracellular loops before activation. These interactions may limit H/D exchange at ECL2 and the exodomain. Histidine H/D exchange allows analysis of site specific changes in protein structure using the imidazole C2-hydrogen on the histidine residue as a probe.65-67 Our histidine H/D exchange analysis of PAR4 showed a twofold increase in the t1/2 for H/D exchange at His229 in thrombin-activated PAR4, which supports a role for ECL2 in PAR4 activation.24 Taken together, these data suggest a more complex arrangement on the extracellular face of PAR4, which can also potentially have an impact on the interactions between PAR4 and thrombin. The conventional model is that proteases interact solely with exodomains of PARs.8,9,14,68,69 Sánchez Centellas et al70 reported that an anionic cluster (Asp224, Asp230, Asp235) in ECL2 is important for PAR4 activation by thrombin, which agrees with a more complex model of PAR4 activation that has emerged with roles for the extracellular loops of the PAR4 and exosite II of thrombin.70-72 Our model in which the N terminus is docked near the extracellular loops would facilitate these additional interactions between PAR4 and thrombin.
The H/D exchange and molecular modeling identified 2 regions of PAR4 that may be important for activation: Thr153 in the LBS and ECL3. Mutating Thr153 to Ala reduced PAR4-mediated Ca2+ signaling. The more conserved substitution to Ser also led to a decrease in Ca2+ mobilization, albeit to a lesser extent (Figure 5; supplemental Figure 3). Taken together, these data demonstrate that both the size and polarity of Thr153 in the LBS are critical for PAR4 signaling. ECL3 had increased H/D exchange in the thrombin-activated PAR4 and adopted an outward confirmation in the computational model. This led us to propose that ECL3 serves as a gatekeeper to block the interaction between the N terminus and the LBS before enzymatic cleavage (Figure 7). In this model, ECL3 swings outward to allow the tethered ligand access to the LBS. We propose a coordinated movement of ECL3 that is facilitated by its rigidity conferred by 2 conserved prolines: Pro310 and Pro312 (Figure 6; supplemental Figure 5).44,73 Mutating either of these residues had an impact on PAR4 signaling, whereas mutating Ser311, which is not predicted to influence loop rigidity, had no effect on signaling. In other studies, mutating Asp309 did not have an impact on signaling, which gives further support to the role of the prolines and loop rigidity.70
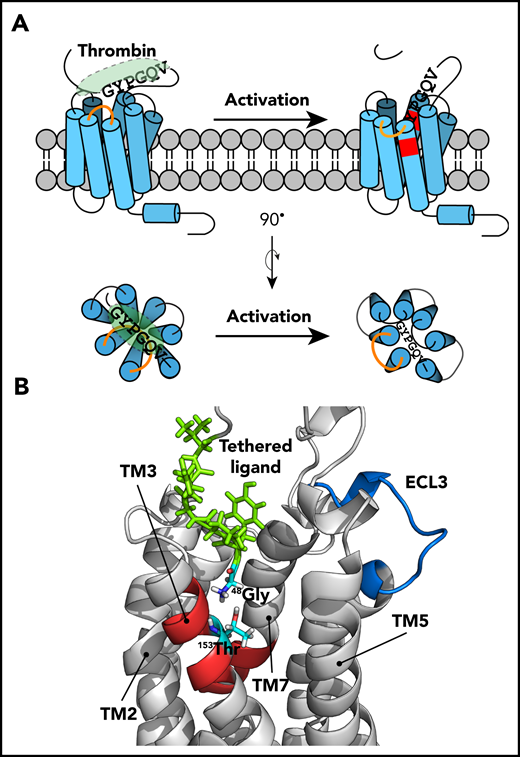
Tethered ligand–mediated PAR4 activation model. (A) Before thrombin activation, the tethered ligand is buried and already docks close to the LBS of PAR4. The interaction between the tethered ligand and its binding site is blocked by ECL3 (orange line). Thrombin cleavage triggers a structural rearrangement of ECL3 that opens the accessibility between the tethered ligand and the binding site. The tethered ligand drops directly into the LBS (shown in red) and the Gly48 interacts with Thr153 to further initiate downstream signaling, which requires G-protein complex to recruit and dock at ICL2 of PAR4. (B) The computational modeling of the thrombin-activated PAR4 model, in which the ECL3 adapted in an “out” position to provide space for the tethered ligand to interact with the binding site further support the working model of the PAR4 activation mechanism. The tethered ligand is green, the LBS is red, and the ECL3 is blue.
Tethered ligand–mediated PAR4 activation model. (A) Before thrombin activation, the tethered ligand is buried and already docks close to the LBS of PAR4. The interaction between the tethered ligand and its binding site is blocked by ECL3 (orange line). Thrombin cleavage triggers a structural rearrangement of ECL3 that opens the accessibility between the tethered ligand and the binding site. The tethered ligand drops directly into the LBS (shown in red) and the Gly48 interacts with Thr153 to further initiate downstream signaling, which requires G-protein complex to recruit and dock at ICL2 of PAR4. (B) The computational modeling of the thrombin-activated PAR4 model, in which the ECL3 adapted in an “out” position to provide space for the tethered ligand to interact with the binding site further support the working model of the PAR4 activation mechanism. The tethered ligand is green, the LBS is red, and the ECL3 is blue.
We observed that PAR4 mutations had greater impact on signaling initiated by PAR4 activating peptide (PAR4-AP) compared with thrombin (Figures 5 and 6). The simple explanation is that the tethered ligand cannot diffuse, which makes it readily available to re-associate with the receptor once the ligand dissociates; this minimizes the impact of the PAR4 mutations. Alternatively, the activation peptides may have distinct interactions and consequences of binding to the receptors. We have recently shown that thrombin-activated PAR4 is resistant to proteolysis by thermolysin, whereas PAR4-AP-stimulated PAR4 had the same digestion profile as unstimulated PAR4.24 These data strongly suggest that the overall conformation is different for the receptor when it is activated by the tethered ligand. Furthermore, signaling initiated by PAR activation peptides is not identical to protease activation of PARs.64,74 Thibeault et al64 recently used an extensive panel of activation peptides to analyze biased signaling from PAR4. They identified specific residues in the PAR4-AP that activated Gq signaling that were distinct from those that activated arrestin signaling. To date, there have been no reports of biased signaling resulting from protease activation of endogenous PAR4. However, Thibeault et al64 and Ramachandran et al75 have shown the capacity of PAR4 to elicit biased signaling. In our studies, we used Ca2+ signaling as a readout of receptor function to validate our H/D exchange and modeling. Going forward, it will be necessary to determine how the regions we identified influence additional signaling pathways downstream of PAR4. In particular, it will be important to determine how the naturally occurring variant PAR4-P310L influences platelet activation via each of these pathways as well as crosstalk with other GPCRs such as P2Y12.50,53,76
A common SNP leads to PAR4 sequence variants at position 120 (Thr or Ala) in TM2 (rs773902). A Thr at this position results in a hyperreactive PAR4 compared with an Ala.13,77,78 To date, the mechanism for this functional difference has not been described, and it is unclear how changing an amino acid in TM2 impacts PAR4 reactivity. Interestingly, Thr120 is near the region we identified as the LBS (supplemental Figure 6). This raises the intriguing possibility that the Thr or Ala at 120 is close enough to the LBS to have an impact on how the tethered ligand interacts with this region. In this study, we also describe the first functional consequences of PAR4 variants at 310 (Pro or Leu) resulting from rs2227376 and directly demonstrate a role for ECL3 and Pro310 in PAR4 activation.
Venous thrombosis and its major complication pulmonary embolism is called VTE and is a major cause of morbidity and mortality.79 Hypercoagulation, stasis, and endothelial dysfunction (Virchow’s triad) are the primary drivers of VTE. Platelets also have an essential initiating role, but the mechanisms are poorly understood.2 Given that PAR4 promotes procoagulant platelets, phosphatidyl serine exposure, and subsequent thrombin generation, it is a potential contributor to VTE.80-82 We evaluated the correlation of 2 PAR4 SNPs, rs773902 and rs2227376, with risk of VTE by using the INVENT Consortium resources.45 Interestingly, PAR4-120A/T (rs773902) was not associated with VTE, whereas the hyporeactive PAR4-310L (rs2227376) was associated with a 15% reduction in relative risk. The differences between these polymorphisms is likely related to their impact on PAR4 reactivity. PAR4-120A has a higher concentration of drug that gives half-maximal response (EC50) than PAR4-120T; however, the maximum signaling for these is the same. In contrast, PAR4-310L has a reduced response to low thrombin concentrations and a lower maximum reactivity, which likely leads to the protection from VTE. The data suggest a previously unrecognized role for PAR4 in the pathology of VTE. Going forward, mechanistic studies are required to determine how PAR4 on platelets and other cells contributes to the initiation and propagation of VTE.
PARs have become promising therapeutic targets.48,83,84 Competing with the tethered ligand creates unique challenges to developing therapeutic agents with favorable pharmacologic profiles. There has been progress with PAR4, but the lack of studies that directly determine the binding site for the endogenous tethered ligand has slowed efforts to development new drugs.12,85 Therefore, high-resolution structure of full-length PAR4 in its natural lipid environment without sequence modifications will greatly facilitate drug discovery. Future studies on PAR4 that combine amide-H/D exchange with recent technological breakthroughs in cryo electron microscopy will untangle the questions raised in our studies. First, a high-resolution structure of activated PAR4 will determine whether Thr153 directly interacts with the tethered ligand or if it merely influences the size and shape of the binding pocket. Second, these will also determine the structural basis for how each SNP contributes to PAR4 function. Third, determining the structure of PAR4 in complex with thrombin will reveal the contributions of ECL2 and ECL3. Fourth, structures of PAR4 signaling complexes in their natural lipid environment will elucidate the mechanism of dimer-mediated signaling.86,87
In conclusion, our findings provide the first description of the PAR tethered ligand activation mechanism. By using H/D exchange, we identified the endogenous tethered LBS and a role for ECL3 in PAR4 activation. We also identified a functional consequence of a PAR4 SNP in ECL3 that leads to a 15% reduction in relative risk of VTE. These studies point to a role for PAR4 in the development of VTE.
Original data is available by e-mailing the corresponding author.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Tivadar Orban for helpful discussions on H/D exchange and critical reading of the manuscript and Nicholas L. Smith for critical reading of the INVENT data.
This study was supported by a grant from the National Institutes of Health (NIH) National Heart Lung and Blood Institute (NHLBI) (HL098217) (M.T.N.) and the American Heart Association (15GRNT25090222) (M.T.N.). X.H. received research funding from the American Heart Association 2018 Predoctoral Fellowship (18PRE33960396) and is cofunded by the Schwab Charitable Fund. This research was supported in part by grant R24EY027283 to K.P. from the NIH National Eye Institute and the Immune Core Facility in the CWRU/UH Center for AIDS Research (NIH grant: P30 AI036219). The authors also acknowledge support from an RPB unrestricted grant to the Department of Ophthalmology, University of California, Irvine. The INVENT Consortium is supported in part by NIH, NHLBI grant HL134894.
Authorship
Contribution: X.H., K.P., and M.T.N. conceived the study; X.H., L.H., M.d.l.F., and M.T.N. designed the experiments; X.H., M.d.l.F., N.A., and M.T.N. performed the experiments and analyzed the data; X.H. and M.T.N. wrote the manuscript; and M.d.l.F., L.H., N.A., and K.P. critically read and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the INVENT Consortium appears in the supplemental appendix.
Correspondence: Marvin T. Nieman, Department of Pharmacology, Case Western Reserve University, 2109 Adelbert Rd, Wood Building, W309B, Cleveland, OH 44106-4965; e-mail: nieman@case.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal