

In this issue Blood, demonstrate for the first time the structural underpinnings for protease-activated receptor 4 (PAR4) activation in the platelet and the role of PAR4 activation in venous thromboembolism (VTE).1
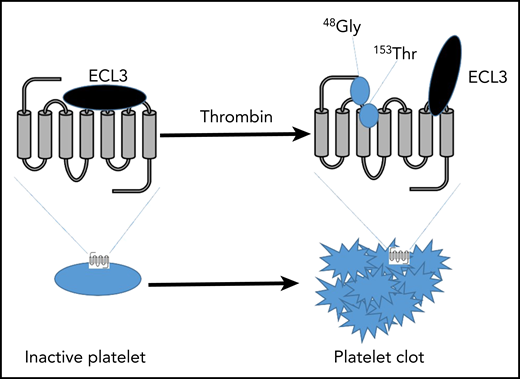
A key aspect PAR4 conformation in inactive platelets is ECL3 functioning as a gatekeeper, inhibiting the tethered ligand from binding to the transmembrane (TM) 3. Upon thrombin stimulation, 48Gly, on the tethered ligand, binds to 153Thr on TM3. 153Thr of TM3, TM7, and 48Gly of the tethered ligand result in receptor activation. PAR4 is now active and induces platelet activation and in vivo clot formation.
A key aspect PAR4 conformation in inactive platelets is ECL3 functioning as a gatekeeper, inhibiting the tethered ligand from binding to the transmembrane (TM) 3. Upon thrombin stimulation, 48Gly, on the tethered ligand, binds to 153Thr on TM3. 153Thr of TM3, TM7, and 48Gly of the tethered ligand result in receptor activation. PAR4 is now active and induces platelet activation and in vivo clot formation.
Platelets express a number of receptors on the surface in order to respond to the changing environment in the blood and vessels. The diversity of receptors expressed on the platelet includes integrin receptors, adhesion receptors, immune receptors, and G protein–coupled receptors (GPCRs). Traditionally, the GPCR class of receptors on the platelet respond to an extremely diverse repertoire of agonists, including prostaglandins, eicosanoids, and small molecules, all of which function to translate a cue in the environment to a response in the cell. Although the diversity of receptors allows for the platelet to engage a variety of responses in different vascular environments, one of the primary functions of the platelet is to respond to inflammatory signals in the blood or at the vessel wall in order to initiate a clot at the site of injury to maintain hemostasis. To this end, the thrombin receptors are thought to be the key signal transducing a coagulation event in the blood to a response by the platelet.
Thrombin receptors, discovered in 1991, are a unique class of GPCRs that are not directly activated by the binding of a ligand in the blood, but rather are activated when a protease in the blood, such as thrombin, binds to the amino terminus and enzymatically cleaves the extracellular tail of the receptor, revealing a new amino-terminal tail that acts as a tethered ligand. Hence, this class of GPCRs, termed protease-activated receptors (PARs), is irreversibly active following exposure to thrombin.2 Four members of the PAR family have been identified to date, with the last receptor, PAR4, being identified in 1998 and being primarily expressed on the platelets in blood.3
Although the first PAR was identified almost 30 years ago, the development of inhibitors targeting the various PARs has been challenging due to the nature of the tethered ligand and its affinity to its activation site on the receptor. With that said, it has been clearly established that PAR activation in the platelet is a key regulator of hemostasis and thrombosis and that inhibition of these receptors might be essential in preventing occlusive thrombotic events in the vessel.4 Since the human platelet expresses both PAR1 and PAR4, understanding their regulation and therapeutic potential has been a focal point of many academic and pharmaceutical research laboratories. Although PAR1 and PAR4 are functionally similar, they are not structurally homologous, and the activation site for PAR4 has remained elusive until now. The focus of the paper by Han et al is to identify for the first time how PAR4 is activated at a structural level and to determine how a specific polymorphism in PAR4 elicits a reduced susceptibility to VTE.
Utilizing a combination of hydrogen exchange and modeling approaches, Han et al were able to exquisitely demonstrate that the tethered ligand is likely predocked in the vicinity of the extracellular loop prior to thrombin cleavage. Furthermore, they were able to present convincing evidence that extracellular loop 3 (ECL3) on PAR4 may function as a gatekeeper to the activation site, preventing the tethered ligand from binding prior to thrombin activation. Furthermore, their data suggest that Thr153 of the ligand binding site directly interacts with Gly48 on the tethered ligand following thrombin activation and that Thr153 binding is critical for the PAR4 ligand to bind and activate the receptor (see figure).
An additional observation uncovered in this study focused on a specific polymorphism endogenously expressed in PAR4 that alters structure and function of the receptor. Although this is not the first polymorphism in PAR4 shown to alter PAR4 function in the platelet,5-7 Leucine at amino acid 310 is the first polymorphism identified in PAR4 that decreases activity. This is a significant discovery as the P310L SNP was then shown through evaluation of a genome-wide association study for VTE to be associated with a decrease in VTE, suggesting this might be a future target for decreasing risk of VTE in patients at risk for deep vein thrombosis.
Overall, this work is a significant advancement in our understanding of the structural regulation of PAR4 activation. Elucidation of its activation site and the ECL3 regulatory function may serve as a blueprint for the development of highly potent and selective inhibitors of PAR4 activation in patients at high risk for thrombosis. Furthermore, identifying a single-nucleotide polymorphism associated with PAR4 and VTE establishes the importance of platelets in this pathology and may function as an important tool for screening patients at potential risk for VTE.
Conflict-of-interest disclosure: The author declares no competing financial interests.
