

Key Points
CK2 and IKAROS regulate chemoresistance to doxorubicin via repression of BCL2L1 (BCL-XL).
Combination treatment with CK2 inhibitor and doxorubicin have a synergistic therapeutic effect on high-risk B-ALL in vivo.

Abstract
High-risk B-cell acute lymphoblastic leukemia (B-ALL) is an aggressive disease, often characterized by resistance to chemotherapy. A frequent feature of high-risk B-ALL is loss of function of the IKAROS (encoded by the IKZF1 gene) tumor suppressor. Here, we report that IKAROS regulates expression of the BCL2L1 gene (encodes the BCL-XL protein) in human B-ALL. Gain-of-function and loss-of-function experiments demonstrate that IKAROS binds to the BCL2L1 promoter, recruits histone deacetylase HDAC1, and represses BCL2L1 expression via chromatin remodeling. In leukemia, IKAROS’ function is impaired by oncogenic casein kinase II (CK2), which is overexpressed in B-ALL. Phosphorylation by CK2 reduces IKAROS binding and recruitment of HDAC1 to the BCL2L1 promoter. This results in a loss of IKAROS-mediated repression of BCL2L1 and increased expression of BCL-XL. Increased expression of BCL-XL and/or CK2, as well as reduced IKAROS expression, are associated with resistance to doxorubicin treatment. Molecular and pharmacological inhibition of CK2 with a specific inhibitor CX-4945, increases binding of IKAROS to the BCL2L1 promoter and enhances IKAROS-mediated repression of BCL2L1 in B-ALL. Treatment with CX-4945 increases sensitivity to doxorubicin in B-ALL, and reverses resistance to doxorubicin in multidrug-resistant B-ALL. Combination treatment with CX-4945 and doxorubicin show synergistic therapeutic effects in vitro and in preclinical models of high-risk B-ALL. Results reveal a novel signaling network that regulates chemoresistance in leukemia. These data lay the groundwork for clinical testing of a rationally designed, targeted therapy that combines the CK2 inhibitor, CX-4945, with doxorubicin for the treatment of hematopoietic malignancies.
Introduction
High-risk B-cell acute lymphoblastic leukemia (B-ALL) in children is often associated with resistance to chemotherapy and an increase in relapse rate.1-6 In adults, B-ALL progresses very aggressively, is often resistant to treatment, and has a poor outcome.7-10 To improve survival, understanding the mechanisms of drug resistance in B-ALL is essential.3 A frequent feature of high-risk B-ALL is the deletion of a single copy of the IKZF1 (IKAROS) gene.2,11-25 IKZF1 encodes a DNA-binding zinc finger protein that functions as a tumor suppressor.26-30 Loss of IKAROS activity in high-risk B-ALL is associated with resistance to conventional chemotherapy and a poor prognosis.11-13,15,16,20,23,31-34 IKAROS binds to the promoters and/or upstream regulatory sequences of its target genes and regulates their transcription via chromatin remodeling.35-43 Despite extensive studies of IKAROS function in leukemia, many questions remain regarding its role in drug resistance. Previous studies have shown that IKAROS’ tumor suppressor function can be impaired by oncogenic casein kinase II (CK2),44-46 an enzyme that is overexpressed in leukemia.47-49 CK2 interferes with IKAROS’ ability to repress cell-cycle progression and the PI3K pathway.49
Here, we present evidence that IKAROS can regulate drug resistance by transcriptionally repressing the BCL2L1 (BCL2-LIKE 1, encodes BCL-XL protein) gene in human B-ALL. In leukemia, phosphorylation of IKAROS by CK2 impairs IKAROS’ function as a transcriptional repressor of BCL2L1. Inhibition of CK2 restores IKAROS-mediated repression of BCL2L1 and increases the sensitivity of leukemia cells to doxorubicin. These results support a new model for the regulation of drug resistance and suggest that CK2 inhibitors can potentiate the therapeutic activity of doxorubicin in hematopoietic malignancies.
Materials and methods
Cell culture and reagents
The Nalm6 B-ALL cell line has been described previously.50 The HEK-293T (293T), JM1, and the N6/ADR, doxorubicin resistance B-ALL cell line were from the American Type Culture Collection (Rockville, MD), and the 697 (EU-3) and REH cell lines were from DSMZ (Braunschweig, Germany).51 Primary human B-ALL cells were cultured as described previously.49,52 IKZF1 mutational status is wild type for cell lines and is shown in supplemental Table 1, available on the Blood Web site, for primary ALL. A summary of patients’ characteristics is in supplemental Table 1. Details regarding reagents and additional experimental methods are found in the supplemental Materials.
Approval for animal studies and use of patient samples
All animal experiments were conducted in the Developmental Therapeutics Preclinical Core facility at Penn State University College of Medicine under protocols approved by the Institutional Animal Care and Use Committee at Penn State University College of Medicine (Hershey, PA). Deidentified patient samples were provided by Loma Linda University (Loma Linda, CA) and the University of Southern California Norris Comprehensive Cancer Center (Los Angeles, CA) in compliance with institutional review board regulations.
Results
IKAROS represses BCL2L1 messenger RNA levels
Analysis of IKAROS’ global genome-wide occupancy in the human B-ALL cell line, Nalm6, 697, and REH, and in primary cells from 2 patients with B-ALL, showed that IKAROS binds to the upstream regulatory element (URE) of the BCL2L1 gene (Figure 1A; supplemental Figures 1 and 2). IKAROS binding at the BCL2L1 URE was determined by a quantitative ChIP (qChIP) assay of primary B-ALL cells from leukemia patients (Figure 1B), and leukemia cell lines and primary leukemia cells from different leukemia subtypes (supplemental Figure 3). IKAROS binding at the BCL2L1 URE was not detected in primary B-ALL with IKZF1 haploinsufficiency caused by deletion of 1 IKZF1 allele Figure 1B (patient 1) nor in embryonic kidney cell carcinoma 293T cells, which do not express IKAROS (supplemental Figure 3).
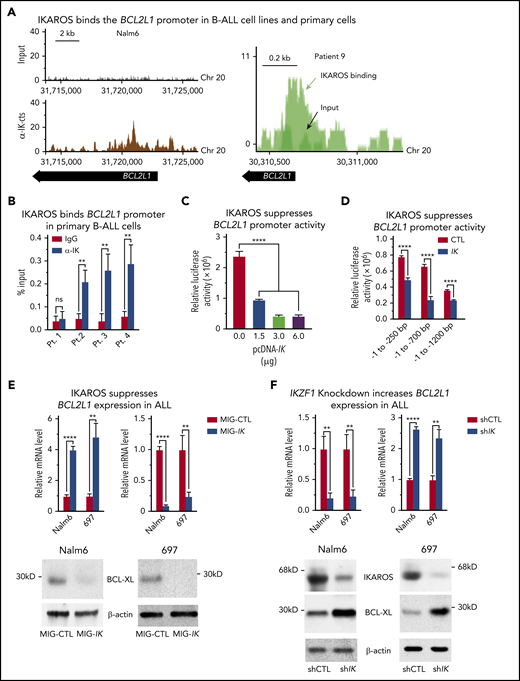
IKAROS binds to the promoter of the BCL2L1 (BCL-XL) gene and suppresses BCL-XL expression. (A) IKAROS binding sites were identified by ChIP-seq in the promoter of BCL2L1 (BCL-XL) in the Nalm6 B-ALL cell line (left) and in a B-ALL patient sample (right). The ChIP-seq data for Nalm6 is analyzed with reference genome MRCh38; the custom tracks are shown on UCSC Genome Browser. The ChIP-seq data for patient 9 is analyzed with reference genome MRCh37 and the custom tracks are shown on CisGenome Browser. (B) The qChIP data confirming IKAROS binding at the BCL2L1 promoter in primary B-ALL cells with wild-type IKZF1 (patients 2-4) but not in IKZF1 haploinsufficiency (patient 1). ChIP enrichments are normalized to input. (C) Activity of the BCL2L1 promoter (located at −1 to −700 bp upstream of transcription start site) was assessed by luciferase reporter assay following transfection with the indicated amount of IKZF1 plasmids (pcDNA-IK) or control vector in 293T cells. (D) IKAROS effect on BCL2L1 promoter activity was assessed by luciferase reporter assay using different regions of the promoter. (E) Nalm6 and 697 B-ALL cell lines were transduced to express IKZF1 (Mig-IK) or with empty vector (MIG-CTL). Relative expression of IKZF1 (left) and BCL-XL (right) assessed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) is graphed. BCL-XL protein levels in Nalm6 and 697 cells were accessed by western blot with anti-BCL-XL specific antibodies (bottom). (F) Nalm6 and 697 B-ALL cell lines were treated with IKZF1 shRNA (shIK) or scramble shRNA control (shCTL). The relative expressions of IKZF1 (left) and BCL2L1 (right) assessed by qRT-PCR are graphed. BCL-XL protein levels in Nalm6 and 697 cells were accessed by western blot with anti-BCL-XL specific antibodies (bottom). (E-F), cells were treated for 3 days. Graphed in panel B is the mean ± SD of 3 independent experiments. Graphed in panels C-F is the mean ± SD of triplicates representative of 1 of 3 independent experiments. **P < .01, ****P < .0001. ns, not significant; pcDNA, plasmid cytomegalovirus DNA; SD, standard deviation.
IKAROS binds to the promoter of the BCL2L1 (BCL-XL) gene and suppresses BCL-XL expression. (A) IKAROS binding sites were identified by ChIP-seq in the promoter of BCL2L1 (BCL-XL) in the Nalm6 B-ALL cell line (left) and in a B-ALL patient sample (right). The ChIP-seq data for Nalm6 is analyzed with reference genome MRCh38; the custom tracks are shown on UCSC Genome Browser. The ChIP-seq data for patient 9 is analyzed with reference genome MRCh37 and the custom tracks are shown on CisGenome Browser. (B) The qChIP data confirming IKAROS binding at the BCL2L1 promoter in primary B-ALL cells with wild-type IKZF1 (patients 2-4) but not in IKZF1 haploinsufficiency (patient 1). ChIP enrichments are normalized to input. (C) Activity of the BCL2L1 promoter (located at −1 to −700 bp upstream of transcription start site) was assessed by luciferase reporter assay following transfection with the indicated amount of IKZF1 plasmids (pcDNA-IK) or control vector in 293T cells. (D) IKAROS effect on BCL2L1 promoter activity was assessed by luciferase reporter assay using different regions of the promoter. (E) Nalm6 and 697 B-ALL cell lines were transduced to express IKZF1 (Mig-IK) or with empty vector (MIG-CTL). Relative expression of IKZF1 (left) and BCL-XL (right) assessed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) is graphed. BCL-XL protein levels in Nalm6 and 697 cells were accessed by western blot with anti-BCL-XL specific antibodies (bottom). (F) Nalm6 and 697 B-ALL cell lines were treated with IKZF1 shRNA (shIK) or scramble shRNA control (shCTL). The relative expressions of IKZF1 (left) and BCL2L1 (right) assessed by qRT-PCR are graphed. BCL-XL protein levels in Nalm6 and 697 cells were accessed by western blot with anti-BCL-XL specific antibodies (bottom). (E-F), cells were treated for 3 days. Graphed in panel B is the mean ± SD of 3 independent experiments. Graphed in panels C-F is the mean ± SD of triplicates representative of 1 of 3 independent experiments. **P < .01, ****P < .0001. ns, not significant; pcDNA, plasmid cytomegalovirus DNA; SD, standard deviation.
We studied the effect of IKAROS expression on BCL2L1 expression using a transient cotransfection assay with various lengths of the BCL2L1 URE (250, 700, and 1200 bp). Cotransfection with IKZF1 resulted in decreased luciferase activity (Figure 1C). This effect was evident even when 250 bp of the BCL2L1 URE was present, but it was most prominent when a region containing 700 bp of the BCL2L1 URE was fused to the reporter gene (Figure 1D). These results suggest that IKAROS acts as a direct repressor of the BCL2L1 promoter and that it binds multiple sites in the BCL2L1 URE.
Next, we tested the effect of increased IKAROS expression on BCL2L1 messenger RNA (mRNA) levels in B-ALL. We compared BCL-XL expression in human Nalm6 and 697 pre-B ALL cells transduced with retrovirus containing wild-type IKZF1 or with empty retroviral vector (negative control). Increased IKZF1 expression resulted in increased IKAROS occupancy at the BCL2L1 promoter (supplemental Figure 4A) as well as reduced BCL2L1 mRNA and BCL-XL protein levels (Figure 1E).
To study the effect of reduced IKAROS activity on BCL2L1 mRNA levels, we targeted IKAROS with short hairpin RNA (shRNA) in Nalm6 and 697 pre–B-all cells. Cells with shRNA-induced IKZF1 knockdown showed a loss of IKAROS binding to the BCL2L1 promoter (supplemental Figure 4B), as well as increased BCL2L1 mRNA and protein levels (Figure 1F). Taken together, these results provide evidence that IKAROS acts as a transcriptional repressor of BCL2L1 in B-ALL.
IKAROS represses BCL2L1 mRNA levels via recruitment of HDAC1
Next, we studied the mechanism by which IKAROS represses BCL2L1. IKAROS can recruit histone deacetylase HDAC1 to the promoters of its target genes, resulting in the alteration of their epigenetic signature and transcriptional repression.41,42 ChIP-seq analysis showed HDAC1 occupancy at the BCL2L1 promoter in Nalm6 cells (Figure 2A). We tested whether IKAROS-mediated repression of BCL2L1 requires histone deacetylase function. The transient cotransfection assay with IKZF1 and luciferase reporter vector fused to the BCL2L1 promoter described here was performed in the presence of a pan-HDAC inhibitor, trichostatin (TSA), or HDAC1 shRNA. Results showed that both TSA and HDAC1 shRNA abolish IKAROS-mediated repression of the BLC2L1 promoter activity (Figure 2B). These data suggest that HDAC1 activity is essential for IKAROS-mediated repression of BCL2L1.
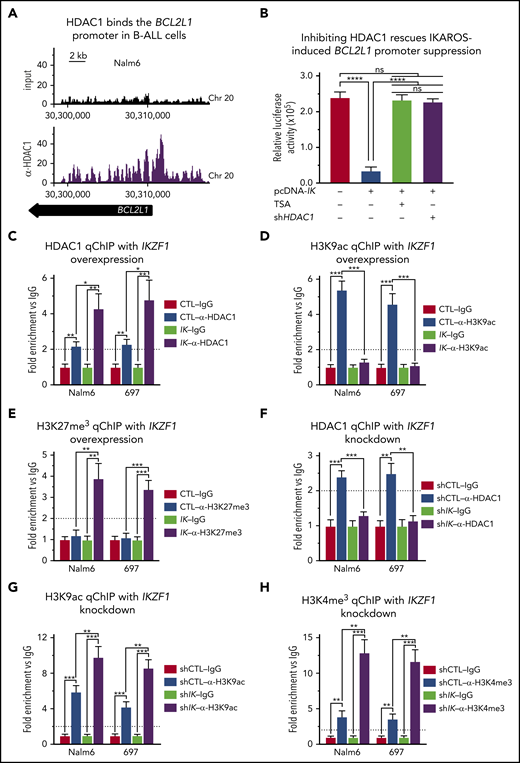
IKAROS represses expression of BCL2L1 (BCL-XL) mRNA through recruitment of HDAC1. (A) HDAC1 binding sites at the BCL2L1 (BCL-XL) promoter were identified by ChIP-seq in Nalm6 cells. ChIP-seq data were analyzed with reference genome MRCh37 and the custom tracks are shown on CisGenome Browser. (B) IKAROS effects on the activity of the BCL2L1 promoter (−1 to −700 bp) were assessed by luciferase reporter assay in the presence of pan-HDACs inhibitor (TSA) or HDAC1 shRNA knockdown (shHDAC1). (C-E) The qChIP data showing binding of HDAC1 (C) and the presence of H3K9ac (D) and H3K27me3 (E) marks at the BCL2L1 promoter in Nalm6 and 697 B-ALL cells with IKAROS (IK) overexpression. (F-H) qChIP data showing binding of HDAC1 (F), H3K9ac (G), and H3K27me3 (H) at the BCL2L1 promoter in Nalm6 and 697 B-ALL cells with IKZF1 shRNA knockdown (shIK). ChIP enrichments are normalized to CTL-IgG samples. Cells were treated for 3 days; graphed data are the mean ± SD of triplicates representative of 1 of 4 independent experiments (C-H). *P < .05, **P < .01, ***P < .001, ****P < .0001. Ig, immunoglobulin.
IKAROS represses expression of BCL2L1 (BCL-XL) mRNA through recruitment of HDAC1. (A) HDAC1 binding sites at the BCL2L1 (BCL-XL) promoter were identified by ChIP-seq in Nalm6 cells. ChIP-seq data were analyzed with reference genome MRCh37 and the custom tracks are shown on CisGenome Browser. (B) IKAROS effects on the activity of the BCL2L1 promoter (−1 to −700 bp) were assessed by luciferase reporter assay in the presence of pan-HDACs inhibitor (TSA) or HDAC1 shRNA knockdown (shHDAC1). (C-E) The qChIP data showing binding of HDAC1 (C) and the presence of H3K9ac (D) and H3K27me3 (E) marks at the BCL2L1 promoter in Nalm6 and 697 B-ALL cells with IKAROS (IK) overexpression. (F-H) qChIP data showing binding of HDAC1 (F), H3K9ac (G), and H3K27me3 (H) at the BCL2L1 promoter in Nalm6 and 697 B-ALL cells with IKZF1 shRNA knockdown (shIK). ChIP enrichments are normalized to CTL-IgG samples. Cells were treated for 3 days; graphed data are the mean ± SD of triplicates representative of 1 of 4 independent experiments (C-H). *P < .05, **P < .01, ***P < .001, ****P < .0001. Ig, immunoglobulin.
We tested whether IKAROS recruits HDAC1 and alters the epigenetic signature of the BCL2L1 promoter in B-ALL using gain-of-function and loss-of function experiments in Nalm6 and 697 cells. Overexpression of IKAROS results in increased HDAC1 occupancy at the BCL2L1 promoter (Figure 2C). This was associated with a loss of H3K9ac enrichment (Figure 2D) and increased enrichment of H3K27me3 (Figure 2E) at the BCL2L1 promoter. Targeting IKZF1 with shRNA resulted in the loss of HDAC1 occupancy at the BCL2L1 promoter (Figure 2F), and increased H3K9ac enrichment, as well as H3K4me3 enrichment at the BCL2L1 promoter (Figure 2G-H). These data suggest that IKAROS represses BCL2L1 mRNA levels via recruitment of HDAC1 and formation of repressive chromatin at the BCL2L1 promoter.
CK2 inhibits IKAROS-mediated repression of BCL2L1
Our next goal was to determine the signaling pathway that controls IKAROS’ ability to regulate BCL2L1 mRNA levels. Previously published data demonstrated that IKAROS is directly phosphorylated by CK2 and that phosphorylation of IKAROS by CK2 impairs its ability to function as a transcriptional regulator.45,46 CK2 is an oncogenic kinase, which is overexpressed in B-ALL,47 and it has been hypothesized that 1 of CK2’s mechanisms of oncogenic action involves the inhibition of IKAROS activity.53 To define the role of CK2 in regulating IKAROS’ function as a repressor of BCL2L1 mRNA levels, the effect of CK2 inhibition in B-cell leukemia was investigated. Molecular inhibition of CK2, using shRNA targeting the catalytic subunit of the CK2 holoenzyme (CK2α), (Figure 3A), resulted in reduced BCL2L1 mRNA levels (Figure 3B). This was associated with increased binding of IKAROS to the BCL2L1 promoter (Figure 3C). Pharmacological inhibition of CK2 with a specific inhibitor, CX-4945, resulted in a large reduction in BCL2L1 mRNA levels (Fig. 3D) and a loss of BCL-XL protein expression (Figure 3E). This was associated with increased binding of IKAROS to the BCL2L1 promoter (Figure 3F; supplemental Figures 5-8), recruitment of HDAC1, increased H3K27me3 occupancy and loss of H3K9ac occupancy at the BCL2L1 promoter (supplemental Figure 9). Treatment with CX-4945 had the same effect on BCL2L1 mRNA levels and IKAROS binding to the BCL2L1 promoter in primary B-ALL cells from patients with both copies of the IKZF1 gene (Figure 3G-H). The use of another specific pharmacological inhibitor of CK2, TBB, also reduced BCL2L1 mRNA levels and increased IKAROS binding to the BCL2L1 promoter in B-ALL cell lines (supplemental Figure 10A-B).
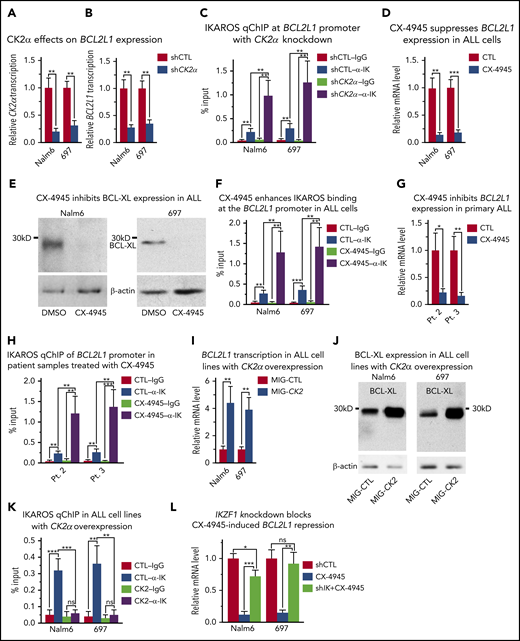
CK2 inhibits IKAROS-mediated repression of BCL2L1 (BCL-XL). (A-B) Effect of CK2α shRNA on mRNA levels of CK2α (A) and BC2L1 (B) in Nalm6 and 697 B-ALL cells. (C) Effect of CK2α shRNA on IKAROS binding at the BC2L1 promoter as measured by qChIP. Effect of pharmacological inhibition of CK2 (with CX-4945) on (D) BCL2L1 mRNA expression level by quantitative reverse transcription polymerase chain reaction (qRT-PCR), (E) BCL-XL protein level by western blot, and (F) IKAROS binding at the BCL2L1 promoter in Nalm6 and 697 B-ALL cells. (G-H) Effect of CX-4945 on the BCL-XL mRNA level (G) and IKAROS binding (H) at the BCL2L1 promoter in primary B-ALL cells. (I-K) Effect of CK2 overexpression (MIG-CK2) and vector only control (MIG-CTL) on BCL2L1 mRNA expression level by qRT-PCR (I), BCL-XL protein level by western blot (J), and IKAROS binding (K) at the BCL2L1 promoter in Nalm6 and 697 B-ALL cells. (L) Effect of IKZF1 knockdown (shIK) on changes in BCL2L1 gene expression induced by CK2 inhibition with CX-4945 as measured by qRT-PCR. Cells were transfected with CK2α in panels A-B or IKZF1 shRNA knockdown in panel K for 3 days using the Neon transfection method, and cells stably expressing lentiviral CK2α in panels I-J are described in the Methods section. (D-H, L) Cells were treated with 5 μM CX-4945 for 2 days. ChIP enrichments are normalized to input. Graphed in panels C, F, H, and K is the mean ± SD of 4 independent experiments. Graphed in panels A-B, D, G, I, L are the mean ± SD of triplicates representative of 1 of 3 independent experiments. *P < .05, **P < .01, ***P < .001.
CK2 inhibits IKAROS-mediated repression of BCL2L1 (BCL-XL). (A-B) Effect of CK2α shRNA on mRNA levels of CK2α (A) and BC2L1 (B) in Nalm6 and 697 B-ALL cells. (C) Effect of CK2α shRNA on IKAROS binding at the BC2L1 promoter as measured by qChIP. Effect of pharmacological inhibition of CK2 (with CX-4945) on (D) BCL2L1 mRNA expression level by quantitative reverse transcription polymerase chain reaction (qRT-PCR), (E) BCL-XL protein level by western blot, and (F) IKAROS binding at the BCL2L1 promoter in Nalm6 and 697 B-ALL cells. (G-H) Effect of CX-4945 on the BCL-XL mRNA level (G) and IKAROS binding (H) at the BCL2L1 promoter in primary B-ALL cells. (I-K) Effect of CK2 overexpression (MIG-CK2) and vector only control (MIG-CTL) on BCL2L1 mRNA expression level by qRT-PCR (I), BCL-XL protein level by western blot (J), and IKAROS binding (K) at the BCL2L1 promoter in Nalm6 and 697 B-ALL cells. (L) Effect of IKZF1 knockdown (shIK) on changes in BCL2L1 gene expression induced by CK2 inhibition with CX-4945 as measured by qRT-PCR. Cells were transfected with CK2α in panels A-B or IKZF1 shRNA knockdown in panel K for 3 days using the Neon transfection method, and cells stably expressing lentiviral CK2α in panels I-J are described in the Methods section. (D-H, L) Cells were treated with 5 μM CX-4945 for 2 days. ChIP enrichments are normalized to input. Graphed in panels C, F, H, and K is the mean ± SD of 4 independent experiments. Graphed in panels A-B, D, G, I, L are the mean ± SD of triplicates representative of 1 of 3 independent experiments. *P < .05, **P < .01, ***P < .001.
Next, we tested the effect of increased CK2 expression on IKAROS’ ability to regulate BCL2L1 gene expression in B-ALL. Nalm6 and 697 cells were transduced with retrovirus that expressed CK2α or an empty vector (as a negative control) and the effect of CK2 overexpression on IKAROS function and BCL2L1 mRNA levels was analyzed. CK2 overexpression results in increased BCL2L1 mRNA levels (Figure 3I), increased BCL-XL protein expression (Figure 3J), and the loss of IKAROS binding to the BCL2L1 promoter (Figure 3K). This was associated with the loss of HDAC1 recruitment to the BCL2L1 promoter and enrichment of H3K4me3 and H3K9ac occupancy at the BCL2L1 promoter (supplemental Figure 11).
These results suggest that CK2 regulates BCL2L1 mRNA levels via IKAROS phosphorylation; however, CK2 phosphorylates more than 200 substrates.54,55 Thus, we tested whether IKAROS is the critical protein through which CK2 regulates BCL-XL expression. Treatment of Nalm6 and 697 cells with the CK2 inhibitor, CX-4945, along with scrambled shRNA, results in reduced BCL2L1 mRNA levels; however, IKZF1 knockdown with shRNA was able to rescue CX-4945-mediated repression of BCL2L1 in both cell lines (Figure 3L). These data show that IKAROS activity is essential for mediating repression of the BCL2L1 gene via CK2 inhibition and suggest that the primary mechanism by which CK2 inhibition represses BCL2L1 expression is through IKAROS.
Together, these data demonstrate that in B-ALL, expression of BCL2L1 (BCL-XL protein) is regulated by the CK2-IKAROS signaling axis, and that alteration in activity of CK2 and/or IKAROS results in changes in BCL2L1 (BCL-XL protein) expression.
CK2 inhibition restores IKAROS’ ability to regulate BCL2L1 (BCL-XL protein) expression in high-risk leukemia
B-cell ALLs that have a deletion or inactivating mutation of 1 IKZF1 allele, are haploinsufficient for IKAROS function. These types of B-ALLs are associated with poor prognosis and often display Ph-like gene expression profiles.1,11 We tested if IKZF1 haploinsufficiency affects IKAROS’ ability to regulate BCL2L1 expression and whether CK2 inhibition can regulate expression of BCL2L1 in IKZF1 haploinsufficient B-ALL cells. We used primary cells from 5 patients with high-risk pediatric B-ALL and deletion of 1 IKZF1 allele (supplemental Table 1). Treatment with CK2 inhibitor resulted in a reduction in BCL2L1 mRNA levels in all 5 primary B-ALL cells (Figure 4A). Analysis of IKAROS binding to the BCL2L1 promoter showed that in primary B-ALL lacking 1 IKZF1 copy, the IKAROS protein does not bind to the BCL2L1 promoter (Figure 4B). Treatment with CK2 inhibitor, CX-4945, restores IKAROS binding in all IKZF1 haploinsufficient B-ALL cells (Figure 4B). We tested whether CK2 inhibition affects IKAROS’ ability to recruit HDAC1 to the BCL2L1 promoter in IKZF1 haploinsufficient high-risk B-ALL. Results showed that in untreated high-risk B-ALL with deletion and/or inactivation of 1 IKZF1 allele, HDAC1 is not recruited to the BCL2L1 promoter (Figure 4C). Treatment with CX-4945 restores recruitment of HDAC1 to the BCL2L1 promoter (Figure 4C). The qChIP analysis of the epigenetic signature at the BCL2L1 promoter showed that all primary high-risk B-ALL cells with deletion/inactivation of 1 IKZF1 allele have an epigenetic signature at the BCL2L1 promoter that is consistent with open/active chromatin. This includes the absence of the repressive epigenetic mark H3K27me3 (Figure 4D), and enrichment of the positive epigenetic marks, H3K9ac (Figure 4E) and H3K4me3 (Figure 4F). Treatment with CK2 inhibitor CX-4945 results in the formation of repressive chromatin characterized by enrichment of H3K27me3 (Figure 4D), and loss of H3K9ac (Figure 4E) and H3K4me3 (Figure 4F) at the BCL2L1 promoter in all 5 high-risk primary B-ALL cells. Altered chromatin remodeling and repressive chromatin formation at the BCL2L1 promoter was also detected when the high-risk B-ALL cells were treated with another CK2 inhibitor, TBB (supplemental Figure 12).
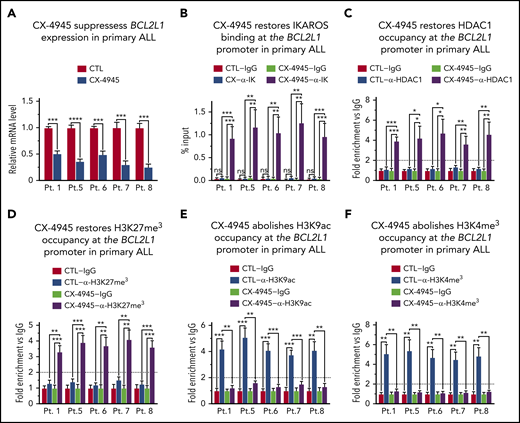
CK2 inhibition restores IKAROS’ ability to regulate BCL2L1 (BCL-XL) expression in primary high-risk B-ALL with deletion of 1 IKZF1 allele. (A) BCL2L1 mRNA level was measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in primary high-risk B-ALL samples with deletion of 1 IKZF1 allele following treatment with 10 μM CK2 inhibitor (CX-4945) for 2 days as compared with untreated (CTRL) cells. The qChIP analysis of the occupancy of (B) IKAROS, (C) HDAC1, (D) H3K27me3, (E) H3K9ac, and (F) H3K4me3 at the BCL2L1 promoter. Untreated cells are (white and light gray bars) are compared with and CX-4945-treated primary high-risk B-ALL (dark gray and black bars). Primary high-risk ALL cells were cultured on stromal cells with or without CK2 inhibitor CX-4945 (10 μM) for 2 days. (B) ChIP enrichments are normalized to input, graphed is mean ± SD of 4 independent experiments. (C-F) ChIP enrichments are normalized to CTL-IgG samples. Graphed in panel A, panels C-F are the mean ± SD of triplicates representative of 1 of 3 independent experiments (A) or 4 independent experiments (C-F). *P < .05, **P < .01, ***P < .001, ****P < .0001.
CK2 inhibition restores IKAROS’ ability to regulate BCL2L1 (BCL-XL) expression in primary high-risk B-ALL with deletion of 1 IKZF1 allele. (A) BCL2L1 mRNA level was measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in primary high-risk B-ALL samples with deletion of 1 IKZF1 allele following treatment with 10 μM CK2 inhibitor (CX-4945) for 2 days as compared with untreated (CTRL) cells. The qChIP analysis of the occupancy of (B) IKAROS, (C) HDAC1, (D) H3K27me3, (E) H3K9ac, and (F) H3K4me3 at the BCL2L1 promoter. Untreated cells are (white and light gray bars) are compared with and CX-4945-treated primary high-risk B-ALL (dark gray and black bars). Primary high-risk ALL cells were cultured on stromal cells with or without CK2 inhibitor CX-4945 (10 μM) for 2 days. (B) ChIP enrichments are normalized to input, graphed is mean ± SD of 4 independent experiments. (C-F) ChIP enrichments are normalized to CTL-IgG samples. Graphed in panel A, panels C-F are the mean ± SD of triplicates representative of 1 of 3 independent experiments (A) or 4 independent experiments (C-F). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Together, these data demonstrate that CK2 inhibition in high-risk B-ALL cells with deletion of 1 IKZF1 allele restores IKAROS’ ability to bind DNA, recruit HDAC1, and form repressive chromatin at the BCL2L1 promoter which results in reduced BCL2L1 mRNA levels.
BCL-XL, IKAROS, and CK2 regulate sensitivity of B-ALL to doxorubicin
BCL-XL is an antiapoptotic protein.56 Increased expression of BCL-XL has been associated with resistance to chemotherapy in T-ALL,57-59 pediatric ALL,60-62 and AML.63-66 We tested the effect of BCL-XL expression on sensitivity to doxorubicin, a drug that is used for standard treatment of high-risk B-ALL. Overexpression of BCL-XL via retroviral transduction, resulted in reduced sensitivity to doxorubicin in B-ALL cell lines (Figure 5A). These data suggest that expression of BCL-XL may mediate resistance to doxorubicin in B-ALL.
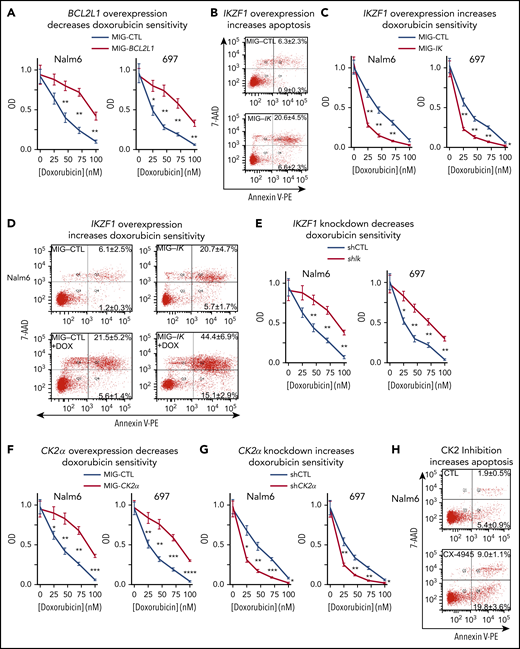
BCL-XL, IKAROS, and CK2 regulate sensitivity to doxorubicin in B-ALL cells. (A) B-ALL cells, retrovirally transduced with BCL2L1 (MIG-BCL2L1) or control vector (MIG-CTL) underwent fluorescence-activated cell sorting (FACS) with the same gate setting of GFP expression, and treated for 3 days with indicated doses of doxorubicin. Cell proliferation was assessed by WST-1 assay. (B) B-ALL cells, retrovirally transduced with IKZF1 or a control vector, were stained with 7-AAD and Annexin V for flow cytometry to determine apoptosis 3 days following retroviral transductions. Gated GFP+ cells were analyzed for apoptosis by flow cytometry. The percentage of cells in the lower right quadrant and upper right quadrant of each flow chart represents the percentage of early apoptotic or late apoptotic cells, respectively, in samples treated with the indicated drugs. (C) B-ALL cells, retrovirally transduced with IKZF1 or a control vector, were FACS-sorted with the same gate setting of GFP expression and treated for 3 days with indicated doses of doxorubicin and assayed using WST-1 cell proliferation assay. (D) B-ALL cells were retroviral transduced with IKZF1 or with control vector and treated with 10 nM doxorubicin (DOX) then evaluated for apoptosis as in panel B. (E) B-ALL cells, transduced with lentiviral IKZF1 shRNA (shIK) or scramble shRNA control (shCTL), underwent FACS with the same gate setting of GFP expression and treated for 3 days with indicated doses of doxorubicin. B-ALL cells with (F) retroviral CK2α overexpression (MIG-CK2α) or vector only control (MIG-CTL); or (G) lentiviral CK2α shRNA or scramble shRNA control (shCTL), underwent FACS based on GFP expression as described previously. FACS cells were treated with indicated doses of doxorubicin for 3 days then evaluated by WST-1 proliferation assay. (H) B-ALL cells were treated with 10 μM CX-4945 for 3 days and stained with 7-AAD and Annexin V for flow cytometry to determine apoptosis. (B, D, H) Flow cytometry plots depict a representative experiment and percentages are the mean ± SD of 3 independent experiments. (A, C, E, F, G) Mean ± SD of triplicates representative of 1 of 3 independent experiments. *P < .05, **P < .01, ***P < .001, ****P < .0001.
BCL-XL, IKAROS, and CK2 regulate sensitivity to doxorubicin in B-ALL cells. (A) B-ALL cells, retrovirally transduced with BCL2L1 (MIG-BCL2L1) or control vector (MIG-CTL) underwent fluorescence-activated cell sorting (FACS) with the same gate setting of GFP expression, and treated for 3 days with indicated doses of doxorubicin. Cell proliferation was assessed by WST-1 assay. (B) B-ALL cells, retrovirally transduced with IKZF1 or a control vector, were stained with 7-AAD and Annexin V for flow cytometry to determine apoptosis 3 days following retroviral transductions. Gated GFP+ cells were analyzed for apoptosis by flow cytometry. The percentage of cells in the lower right quadrant and upper right quadrant of each flow chart represents the percentage of early apoptotic or late apoptotic cells, respectively, in samples treated with the indicated drugs. (C) B-ALL cells, retrovirally transduced with IKZF1 or a control vector, were FACS-sorted with the same gate setting of GFP expression and treated for 3 days with indicated doses of doxorubicin and assayed using WST-1 cell proliferation assay. (D) B-ALL cells were retroviral transduced with IKZF1 or with control vector and treated with 10 nM doxorubicin (DOX) then evaluated for apoptosis as in panel B. (E) B-ALL cells, transduced with lentiviral IKZF1 shRNA (shIK) or scramble shRNA control (shCTL), underwent FACS with the same gate setting of GFP expression and treated for 3 days with indicated doses of doxorubicin. B-ALL cells with (F) retroviral CK2α overexpression (MIG-CK2α) or vector only control (MIG-CTL); or (G) lentiviral CK2α shRNA or scramble shRNA control (shCTL), underwent FACS based on GFP expression as described previously. FACS cells were treated with indicated doses of doxorubicin for 3 days then evaluated by WST-1 proliferation assay. (H) B-ALL cells were treated with 10 μM CX-4945 for 3 days and stained with 7-AAD and Annexin V for flow cytometry to determine apoptosis. (B, D, H) Flow cytometry plots depict a representative experiment and percentages are the mean ± SD of 3 independent experiments. (A, C, E, F, G) Mean ± SD of triplicates representative of 1 of 3 independent experiments. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Loss of IKAROS activity is associated with high-risk leukemia.32,67 One of the features of high-risk leukemia is resistance to chemotherapy.31,67 Because our data demonstrated that IKAROS represses expression of the BCL2L1 gene and that BCL-XL regulates resistance to doxorubicin, we tested the effect of IKAROS expression on sensitivity to doxorubicin treatment in B-ALL. IKAROS overexpression results in increased apoptosis of B-ALL cells (Figure 5B), which correlates with repression of the BCL-XL antiapoptotic protein. B-ALL cells with overexpression of IKAROS have increased sensitivity to doxorubicin treatment (Figure 5C) and increased doxorubicin-induced apoptosis (Figure 5D). Knockdown of IKZF1with shRNA resulted in reduced sensitivity to doxorubicin-induced cytotoxicity (Figure 5E). These results showed that IKAROS expression directly correlates with sensitivity to doxorubicin treatment.
Because CK2 inhibition reduces BCL2L1 mRNA levels via IKAROS, we tested if CK2 inhibition would enhance apoptosis and chemosensitivity to doxorubicin in B-ALL. Results showed that overexpression of CK2α via retroviral transduction results in reduced sensitivity to doxorubicin-induced cytotoxicity in B-ALL cell lines (Figure 5F). Correspondingly, knock-down of CK2α with shRNA results in increased cytotoxicity following treatment with doxorubicin in both Nalm6 and 697 cells (Figure 5G). Treatment of B-ALL cells with the CK2 inhibitor CX-4945 results in increased apoptosis, as evidenced by flow cytometry (Figure 5H).
Overall, the presented data demonstrate that chemosensitivity to doxorubicin is determined by BCL-XL expression, which is regulated by CK2 and IKAROS activity. Results suggest that reduced CK2 activity and/or increased IKAROS function increases chemosensitivity to doxorubicin treatment in B-ALL cells.
CK2 inhibition synergizes with doxorubicin in the treatment of B-ALL cells
Because the results presented in Figure 5 demonstrated a role for CK2 in chemoresistance to doxorubicin, we tested whether the combination of CK2 inhibitor and doxorubicin exerted synergistic therapeutic effects on B-ALL cells. We used the CK2 inhibitor, CX-4945,68 which is currently being tested in a phase 1 trial,55 and compared the therapeutic effect of CX-4945 and doxorubicin combination therapy vs single-drug treatment, in vitro, on 2 different human B-ALL cell lines, Nalm6 and 697. CalcuSyn analyses show that the combination of CX-4945 and doxorubicin, given at doses that are achievable in serum in patients, produced synergistic cytotoxic effects in both Nalm6 (Figure 6A) and 697 (Figure 6B) B-ALL cell lines.
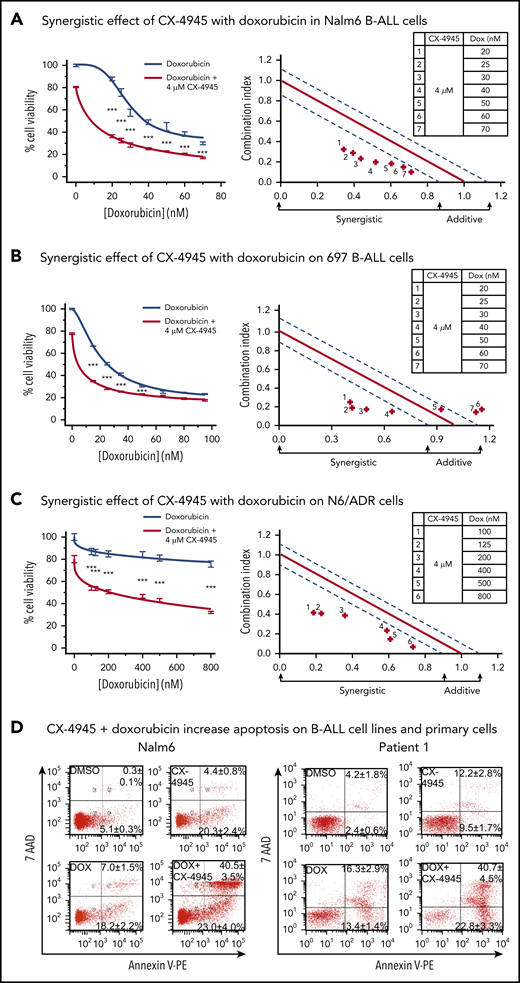
Synergistic effects of CX-4945 with doxorubicin on cell proliferation arrest and apoptosis in B-ALL cells. (A-C) Effects (left) and synergistic analysis (right) of doxorubicin (gray line) and the combination of doxorubicin and CX-4945 (red line) on proliferation of Nalm6 (A) and 697 (B), and drug-resistant N/6 B-ALL (C) cells. Cells were treated with the indicated doses of drugs for 2 days. Cellular proliferation was measured by WST-1 assay. Synergistic analysis was performed with Calcusyn; where combination index value of: 1.15 to 0.8 is considered additive effect; 0.85 to 0.7 is moderately synergistic; and <0.7 is very synergistic effect, respectively. ***P < .001. (D) Effect of CX-4945 (4 μM), doxorubicin (DOX, 3.2 nM), and the combination of doxorubicin (3.2 nM) plus CX-4945 (4 μM) on apoptosis in Nalm6 (left) and patient 1 (right) B-ALL cells. Cells were treated for 2 days and stained with 7-AAD and annexin V for flow cytometry to assess apoptosis. The percentage of cells in the lower right quadrant and upper right quadrant of each flowchart represents the percentage of early apoptotic or late apoptotic cells, respectively, in samples treated with the indicated drugs. Mean ± SD of triplicates representative of 1 of 3 independent experiments (A-C). Flow cytometry plots in panel D depict a representative experiment and percentages are the mean ± SD of 3 independent experiments.
Synergistic effects of CX-4945 with doxorubicin on cell proliferation arrest and apoptosis in B-ALL cells. (A-C) Effects (left) and synergistic analysis (right) of doxorubicin (gray line) and the combination of doxorubicin and CX-4945 (red line) on proliferation of Nalm6 (A) and 697 (B), and drug-resistant N/6 B-ALL (C) cells. Cells were treated with the indicated doses of drugs for 2 days. Cellular proliferation was measured by WST-1 assay. Synergistic analysis was performed with Calcusyn; where combination index value of: 1.15 to 0.8 is considered additive effect; 0.85 to 0.7 is moderately synergistic; and <0.7 is very synergistic effect, respectively. ***P < .001. (D) Effect of CX-4945 (4 μM), doxorubicin (DOX, 3.2 nM), and the combination of doxorubicin (3.2 nM) plus CX-4945 (4 μM) on apoptosis in Nalm6 (left) and patient 1 (right) B-ALL cells. Cells were treated for 2 days and stained with 7-AAD and annexin V for flow cytometry to assess apoptosis. The percentage of cells in the lower right quadrant and upper right quadrant of each flowchart represents the percentage of early apoptotic or late apoptotic cells, respectively, in samples treated with the indicated drugs. Mean ± SD of triplicates representative of 1 of 3 independent experiments (A-C). Flow cytometry plots in panel D depict a representative experiment and percentages are the mean ± SD of 3 independent experiments.
We tested whether the CX-4945 and doxorubicin combination treatment would have a synergistic therapeutic effect on a multidrug-resistant B-ALL cell line. The N6/ADR cell line is a subclone of the Nalm6 cell line that is resistant to doxorubicin, vincristine, and etoposide and expresses P-glycoprotein, which is consistent with the “classical” P-glycoprotein-associated multidrug-resistant phenotype.69,70 Results show that the combination of CX-4945 and doxorubicin had a synergistic cytotoxic effect against multidrug-resistant B-ALL (Figure 6C), which suggests that this combination treatment can be efficacious in chemotherapy-resistant, high-risk B-ALL.
Because the inhibition of CK2 by CX-4945 results in decreased expression of BCL-XL in B-ALL, we tested whether 1 of the mechanisms of the synergistic cytotoxic effects of the CX-4945 and doxorubicin combination treatment involves increased apoptosis in B-ALL cells. Results showed that CX-4945 and doxorubicin, in combination, induced increased apoptosis of Nalm6 B-ALL cells compared with treatment with either drug alone (Figure 6D, left, lower right plot). Additionally, CX-4945 in combination with doxorubicin resulted in augmented apoptosis as compared with treatment with either single drug in primary B-ALL cells (Figure 6D, right, lower right plot).
Overall, these results demonstrate that combination therapy with CX-4945 and doxorubicin have synergistic cytotoxic activity in B-ALL and that 1 of the mechanisms responsible for this synergistic effect involves augmenting the pro-apoptotic effect of doxorubicin via CK2 inhibition.
CK2 inhibition augments cytotoxicity of doxorubicin in PDX of primary high-risk B-ALL
The synergistic cytotoxic effects of CK2 inhibitor, CX-4945, in combination with doxorubicin, in 3 different B-ALL cell lines, led to the hypothesis that CX-4945 could augment the therapeutic activity of doxorubicin against B-ALL in vivo. Treatment of high-risk B-ALL includes anthracyclines (eg, doxorubicin). We tested the therapeutic effect of the CX-4945 and doxorubicin in combination vs single-drug therapy in vivo in preclinical models of B-ALL. We used patient-derived xenograft (PDX) models produced from patients with high-risk B-ALL. B-ALL was determined to be high-risk based on negative prognostic markers or clinical features (supplemental Table 1). B-ALL cells were injected via tail vein into immunodeficient NRG mice. Following engraftment, mice from each PDX were divided into the following 4 treatment groups: group 1 (control): vehicle; group 2: CX-4945 daily at 100 mg/kg per day for 21 days); group 3: doxorubicin 1 mg/kg intraperitoneally once per week; and group 4: CX-4945 and doxorubicin combination treatment using the same doses as provided previously. Following the treatment period, the total living leukemia cells in bone marrow (BM) and spleen of mice was determined by flow cytometry.
Results showed that treatment with CX-4945 or doxorubicin as a single drug has a strong therapeutic effect in vivo in all 4 PDX (Figure 7A-C; supplemental Figures 13-17). This was evidenced by the reduced total number of viable B-ALL cells in both the BM and spleen of treated mice, as compared with the untreated control. Although both drugs had obvious therapeutic effects as a single drug, the strong presence of leukemia in both BM and spleen remained evident, suggesting that treatment with a single drug (CX-4945 or doxorubicin) could not induce remission. Combination treatment that included both CX-4945 and doxorubicin given over the 3-week period produced a significantly stronger therapeutic effect in all 4 PDX models (Figure 7A-C) without significant toxicity (supplemental Table 2). The total number of viable leukemia cells was severely reduced (three- to fourfold) in the BM and spleen of the PDX mice treated with combination therapy compared with mice treated with CX-4945 or doxorubicin alone. These results demonstrate that an inhibitor of oncogenic CK2 kinase (CX-4945) and doxorubicin have synergistic therapeutic and cytotoxic effects on high-risk B-ALL cells, when given as a combination treatment, in vivo.
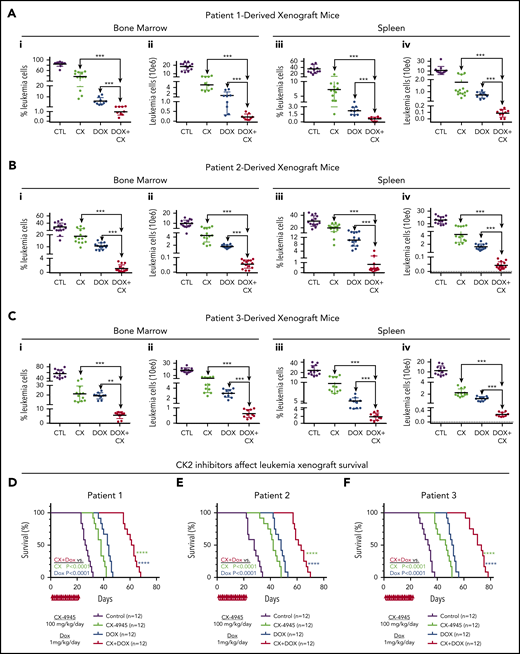
CK2 inhibition augments cytotoxicity of doxorubicin in PDX models of primary high-risk B-ALL. NRG mice were transplanted via tail vein with primary B-ALL cells (2 × 10E6 cells/mouse) from 3 patients. Mice were randomly divided into 4 groups. Once engraftment was established (determined as described in "Methods"), mice were treated with CX4945 (CX) only, doxorubicin (DOX) only, CX+DOX, or with vehicle-only control as described in "Methods." (A-C) Mice were euthanized and evaluated for the presence of leukemia in BM and spleen at day 24 following the initiation of treatment. BM and spleen cells were harvested and counted, and also stained for flow cytometry to detect human B-cell markers (CD10 and CD19), mouse CD45, and 7-AAD as a dead cell marker (described in "Methods"). The percentage of the living B-ALL leukemia cells (i, iii) and total leukemia cells (ii, iv) in BM and spleen were calculated and graphed. The effect of drug treatment was assessed by Student t test. (D-F) Patient-derived xenografts established with B-ALL from patient 1 (D), patient 2 (E), and patient 3 (F) were treated for 22 days with CK2 inhibitors, CX-4945 (CX), doxorubicin (DOX) only, CX+DOX, or vehicle control and followed for survival. Survival curves were generated using the Kaplan-Meier method and differences in survival were analyzed by χ2 test. **P < .01, ***P < .001, ****P < .0001.
CK2 inhibition augments cytotoxicity of doxorubicin in PDX models of primary high-risk B-ALL. NRG mice were transplanted via tail vein with primary B-ALL cells (2 × 10E6 cells/mouse) from 3 patients. Mice were randomly divided into 4 groups. Once engraftment was established (determined as described in "Methods"), mice were treated with CX4945 (CX) only, doxorubicin (DOX) only, CX+DOX, or with vehicle-only control as described in "Methods." (A-C) Mice were euthanized and evaluated for the presence of leukemia in BM and spleen at day 24 following the initiation of treatment. BM and spleen cells were harvested and counted, and also stained for flow cytometry to detect human B-cell markers (CD10 and CD19), mouse CD45, and 7-AAD as a dead cell marker (described in "Methods"). The percentage of the living B-ALL leukemia cells (i, iii) and total leukemia cells (ii, iv) in BM and spleen were calculated and graphed. The effect of drug treatment was assessed by Student t test. (D-F) Patient-derived xenografts established with B-ALL from patient 1 (D), patient 2 (E), and patient 3 (F) were treated for 22 days with CK2 inhibitors, CX-4945 (CX), doxorubicin (DOX) only, CX+DOX, or vehicle control and followed for survival. Survival curves were generated using the Kaplan-Meier method and differences in survival were analyzed by χ2 test. **P < .01, ***P < .001, ****P < .0001.
The effect of CX-4945 treatment on BCL2L1 mRNA levels during in vivo treatment was analyzed before cytotoxicity occurred, at days 3 and 7 following the initiation of in vivo treatment with single drugs or combination therapy with CX-4945 and doxorubicin as described previously. Results showed that in vivo treatment of primary xenografts with the CK2 inhibitor, CX-4945, in combination with doxorubicin, results in reduced BCL2L1 mRNA levels in leukemia cells in both BM and spleen (supplemental Figure 18).
Mice were followed for survival using the Kaplan-Meier method. Results showed that combination treatment with CX-4945 and doxorubicin significantly prolongs survival of mice compared with single-drug treatment (Figure 7D; supplemental Figure 13).
Together, these results demonstrate that combination treatment with the CK2 inhibitor, CX-4945, and doxorubicin has a superior therapeutic effect in high-risk B-ALL preclinical models, compared with single-drug treatment. These results suggest that 1 of the mechanisms through which CK2 inhibition augments the therapeutic effect of doxorubicin in vivo includes reducing BCL2L1 mRNA levels.
We tested whether CX-4945 treatment acts synergistically with dexamethasone or vincristine, 2 drugs commonly used to treat B-ALL. Results showed that the combination of CX-4945 and dexamethasone, as well as CX-4945 and vincristine, exerted synergistic therapeutic effects on B-ALL (supplemental Figure 19).
Discussion
Impaired IKAROS function results in high-risk B-ALL, which is associated with resistance to chemotherapy.11-13,71 Past studies provided evidence that IKAROS can regulate sensitivity to steroid treatment72-74 and to tyrosine kinase inhibitors.6 The data presented here demonstrate that IKAROS regulates drug resistance by altering expression of the antiapoptotic gene BCL2L1. Gain- and loss-of-function experiments showed that IKAROS normally reduces BCL2L1 mRNA levels via recruitment of HDAC1 histone deacetylase and inducing the formation of repressive chromatin. In high-risk leukemia, IKAROS’ function as a BCL2L1 repressor is impaired because of phosphorylation by the oncogenic kinase CK2 that is overexpressed in leukemia. Previously published data show that phosphorylation by CK2 interferes with IKAROS’ ability to regulate a large number of genes, which led to the identification of the CK2-IKAROS signaling axis that regulates cellular proliferation in leukemia.49,75-81 The presented data identify a novel role of CK2-IKAROS signaling: regulation of drug resistance and apoptosis via transcriptional control of BCL2L1 expression.
The role of BCL-XL in cell survival,82,83 malignant transformation,84,85 and chemoresistance,86,87 specifically in context of doxorubicin in leukemia and other malignancies,59,88-90 is well-established. Targeting apoptosis is an attractive therapeutic approach for both hematological and solid tumor malignancies.91-93 Inhibitors of the BCL-XL protein have produced very strong preclinical results,94-96 although clinical application has been hampered by severe thrombocytopenia.97-99 Our results identify a novel way to target apoptosis—repressing transcription of the BCL2L1 gene via inhibition of CK2 and reactivation of IKAROS’ function as a transcriptional repressor. This approach produced a strong synergistic therapeutic effect with doxorubicin in vivo without evidence of thrombocytopenia. Reasons for the absence of thrombocytopenia are unclear (potential explanations include residual BCL-XL expression and/or other effects of CK2 inhibition), but minimal treatment side effects and the synergistic therapeutic effect are promising outcomes for the clinical application of CX-4945 and doxorubicin in combination therapy. The comparable synergistic efficacy of combined CK2 inhibition and doxorubicin in the 2 xenografts without IKZF1 deletion compared with the patient 1 and patient 7-derived xenograft (with Ikaros haploinsufficiency) is likely the result of the functional inactivation of Ikaros caused by overexpression of CK2 in B-ALL in other xenografts.
Importantly, CK2 inhibition restored IKAROS DNA binding and repression of BCL-XL in primary B-ALL cells from patients with partial deletion of 1 IKZF1 allele that results in truncated IKAROS protein, which includes a protein interaction domain and has the potential to act as a “dominant-negative” (Figure 4). This is likely because the truncated IKAROS may reduce, but does not abolish, the DNA binding of IKAROS produced from the remaining wild-type allele (supplemental Figure 20). Combination treatment with CX-4945 and doxorubicin showed a superior therapeutic effect in PDXs derived from patients with B-ALL and truncated IKAROS from 1 allele (Figure 7A, patient 1; supplemental Figure 13, patient 7), supporting the clinical application of this combination treatment of such patients. However, in the rare patients with deletions in both IKZF1 alleles, this combination therapy might be less beneficial.
Doxorubicin, as well as the other anthracycline, mitoxantrone, are extensively used as standard chemotherapy for newly diagnosed and relapsed B-ALL, which makes the clinical relevance of the presented data very high. The prognosis for relapsed pediatric B-ALL has not improved in 30 years, and new therapeutic modalities are needed. These data establish a rationally designed, mechanism-based, targeted therapy that combines the CK2 inhibitor, CX-4945, with doxorubicin as a novel therapeutic approach for the treatment of high-risk and/or relapsed B-ALL (supplemental Figure 21). The synergistic therapeutic effects of CX-4945 with dexamethasone and with vincristine suggest that CK2 inhibition could be effective in combination with these drugs for B-ALL treatment as well. The essential role of BCL-XL for cell survival in most types of T-ALL58,59 suggest that this combination might also be effective in treating T-ALL.
The results of our study establish a new paradigm for targeting chemoresistance: targeting the transcriptional network that regulates expression of the gene encoding the chemoresistant protein, as opposed to targeting the protein itself, and combining this approach with an established chemotherapy agent (supplemental Figure 21). Because the transcriptional regulatory networks of many genes that encode proteins involved in chemoresistance are well-established, this approach opens new possibilities for targeted combination therapies.
In summary, our presented data establish the therapeutic efficacy of a novel combination treatment that targets chemoresistant, high-risk B-ALL in a preclinical model. The approach proposed in the study, targeting the transcriptional regulatory network of genes encoding drug-resistant proteins, combined with standard chemotherapeutic agents, can provide a paradigm for similar targeted combination treatments for drug-resistant hematological malignancies.
ChIP-Seq data are accessible on Gene Expression Omnibus with an access number of GSE44218 at the following link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44218 (Nalm6 data) and at access number GSE58825 at the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? (primary B-ALL data).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from National Institutes of Health, National Cancer Institute R01CA209829 (K.J.P. and S.D.), R01CA213912 (S.D. and C.S.), F30CA221109 (J.L.P.), National Institute of Diabetes and Digestive and Kidney Diseases (R01DK110108) and National Cancer Institute (R01CA204044) (S.H.), and National Institute of General Medical Sciences (R35GM124820) and National Human Genome Research Institute (R01HG009906) (F.Y.); National Center for Advancing Translational Sciences (KL2 TR002015) (C.G.); Hyundai Hope on Wheels Scholar Grant (C.G.); National Natural Science Foundation of China (81770172) (Z.G.); Four Diamonds Fund of the Pennsylvania State University College of Medicine (S.D., C.G., and C.S.); Bear Necessities Pediatric Cancer Foundation; Alex’s Lemonade Stand Foundation (S.D.), John Wawrynovic Leukemia Research Scholar Endowment (S.D. and C.G.); St. Baldrick’s Foundation (S.D. and C.G.); and Rally Foundation (C.G.).
Authorship
Contribution: S.D. analyzed and interpreted data, wrote the manuscript, and designed research; C.G. analyzed and interpreted data and wrote the manuscript; C.S., Z.G., and Y.D. performed research and wrote the manuscript; K.J.P. analyzed and interpreted data and wrote the manuscript; S.A., G.P.R., S.H., V.S., J.A.Y., Y.Y., and M. Muschen analyzed and interpreted data; D.D. collected data and performed statistical analysis; F.Y. and Y.I.K. performed statistical analysis; M.E.R. provided critical review and assisted in writing the manuscript; and B.-H.T., K.G., R.G., J.L.P., Z.G., S.I., P.K.D., M.X., N.M.C., M. McGrath, J.S., R.S., Z.Z. and X.L. performed research.
Conflict-of-interest disclosure: K.J.P. is chief executive officer and owns stock in Elf Zone Inc., a startup company developing therapies for B-cell acute lymphoblastic leukemia. The remaining authors declare no competing financial interests.
Correspondence: Chandrika Gowda, Division of Pediatric Hematology/Oncology, Department of Pediatrics, Pennsylvania State University College of Medicine, 500 University Dr, Mail Code HO85, Hershey, PA 17033; e-mail: cgowda2@pennstatehealth.psu.edu; and Sinisa Dovat, Division of Pediatric Hematology/Oncology, Department of Pediatrics, Pennsylvania State University College of Medicine, 500 University Dr, Mail Code HO85, Hershey, PA 17033; e-mail: sdovat@pennstatehealth.psu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal