

Key Points
Plasma PK, the precursor of the enzyme kallikrein, has intrinsic proteolytic activity.
PK activity may contribute to bradykinin production and to initiation of contact activation when blood is exposed to surfaces.

Abstract
Prekallikrein (PK) is the precursor of the trypsin-like plasma protease kallikrein (PKa), which cleaves kininogens to release bradykinin and converts the protease precursor factor XII (FXII) to the enzyme FXIIa. PK and FXII undergo reciprocal conversion to their active forms (PKa and FXIIa) by a process that is accelerated by a variety of biological and artificial surfaces. The surface-mediated process is referred to as contact activation. Previously, we showed that FXII expresses a low level of proteolytic activity (independently of FXIIa) that may initiate reciprocal activation with PK. The current study was undertaken to determine whether PK expresses similar activity. Recombinant PK that cannot be converted to PKa was prepared by replacing Arg371 with alanine at the activation cleavage site (PK-R371A, or single-chain PK). Despite being constrained to the single-chain precursor form, PK-R371A cleaves high-molecular-weight kininogen (HK) to release bradykinin with a catalytic efficiency ∼1500-fold lower than that of kallikrein cleavage of HK. In the presence of a surface, PK-R371A converts FXII to FXIIa with a specific activity ∼4 orders of magnitude lower than for PKa cleavage of FXII. These results support the notion that activity intrinsic to PK and FXII can initiate reciprocal activation of FXII and PK in solution or on a surface. The findings are consistent with the hypothesis that the putative zymogens of many trypsin-like proteases are actually active proteases, explaining their capacity to undergo processes such as autoactivation and to initiate enzyme cascades.
Introduction
Prekallikrein (PK) is the precursor of kallikrein (plasma protease kallikrein [PKa]),1,2 a plasma protease that cleaves high-molecular-weight kininogen (HK) to release the vasoactive peptide bradykinin (BK),3,4 and contributes to blood clotting in the activated partial thromboplastin time (aPTT) assay. Most PK (70% to 80%) circulates in plasma as a noncovalent complex with HK.5 Reducing plasma PK concentrations in mice and primates reduces BK production.6,7 Deficiency of C1 inhibitor (C1-INH), a major regulator of PKa production and activity, is associated with bouts of soft tissue swelling (angioedema) due to excessive BK production.8-10 These observations support the premise that PKa contributes to setting vascular tone and permeability by regulating basal BK generation.
PK is converted to PKa by proteolysis after Arg371.1,2 Several proteins catalyze or enhance this reaction including the plasma protease factor XIIa (FXIIa),11 the lysosomal enzyme prolyl-carboxypeptidase,12 and the chaperone heat shock protein 90.13 The major PK activator in blood is FXIIa. FXIIa in turn is formed when its precursor, FXII, is cleaved after Arg353 by PKa.11,14,15 Mixing PK and FXII in solution leads to conversion of both to the active forms by a process that is accelerated when the proteins bind to surfaces (contact activation).16-18 Surface enhancement of reciprocal FXII-PK activation contributes to initiation of coagulation in the aPTT assay, and to thrombosis and inflammation when blood is exposed to medical devices such as cardiopulmonary bypass and renal dialysis circuits.19-22
It is postulated that traces of FXIIa or PKa that are always present in plasma initiate FXII-PK activation.23,24 However, we recently showed that FXII expresses a low level of proteolytic activity, independently of FXIIa, that converts PK to PKa.17,25 FXII should, therefore, be considered a protease (an enzyme) rather than a zymogen (an inactive enzyme precursor). It was reported that plasma PK cleaves HK to release BK by a FXII-independent process,26,27 suggesting that PK is also a protease. A potential confounding factor relevant to experiments utilizing proteins derived from plasma is that contamination with activated protease may be partially or entirely responsible for the observed effect. To address this, we prepared a version of PK that cannot be converted to PKa and tested its capacity to cleave the PKa substrates HK and FXII.
Experimental procedures
Materials
Materials were obtained as follows: normal plasma (Precision BioLogic); PK-deficient plasma (George King Biomed); human plasma-derived FXII, FXIIa, PK, and PKa, and corn trypsin inhibitor (CTI) (Enzyme Research Laboratory); C1-INH (Sigma-Aldrich); S-2302 (H-d-prolyl-l-phenylalanyl-l-arginine-p-nitroaniline) (DiaPharma); and PTT-A reagent (Diagnostica Stago). Polyphosphate (poly-P; 60-100 U, not calcium saturated; ILC Performance Products) was a gift from Thomas Renné and Katrin Nickel (University Medical Center, Hamburg, Germany). Genomic DNA was prepared from human leukocytes by phenol-chloroform extraction.
Antibodies
Monoclonal immunoglobulin Gs (IgGs) specific for FXIIa (559C-X181-D06 [D06])17,18 and PKa (559A-M202-H03 [H03])28 are described. Goat polyclonal IgGs (horseradish peroxidase [HRP] conjugated and nonconjugated) against human PK or FXII were obtained from Affinity Biologicals. Goat anti-human HK IgG was obtained from Nordic Immunology.
Expression of recombinant PK, FXII, and FXI
Figure 1A contains diagrams of human PK and its active forms, and human FXII and FXI. PK,1 FXII,14 and factor XI (FXI)29 complementary DNAs were introduced into pJVCMV and expressed in HEK293 cells under serum-free conditions.16,17 In addition to wild-type PK (PK-WT), FXII (FXII-WT), and FXI (FXI-WT), variants were prepared with alanine replacing arginine at the activation cleavage site (PK-R371A and FXI-R369A) or serine in the active site (PK-S559A, FXII-S544A,17 and FXI-S557A).
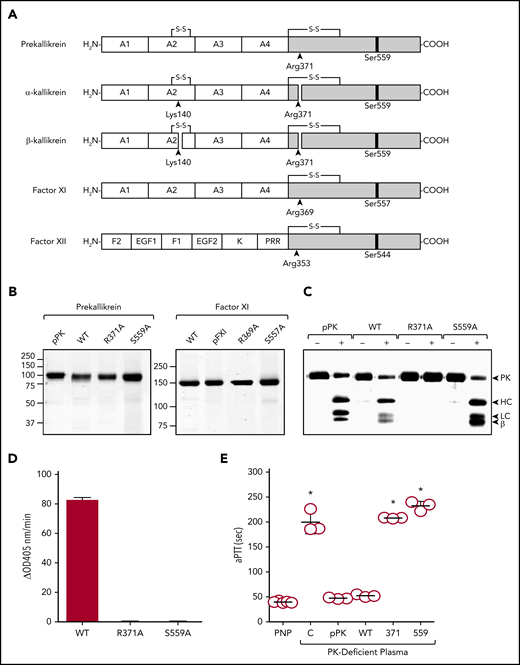
Recombinant proteins. (A) Schematic diagrams of contact activation proteases. Catalytic domains are indicated by gray boxes, and noncatalytic domains by white boxes. Positions of active site serine residues are indicated by black bars. Sites of proteolysis during activation are indicated by arrows, with black arrows indicating sites of cleavage required for full protease activity. PK is an 86-kDa polypeptide that is cleaved after Arg371 to form α-kallikrein (PKa). A second cleavage after Arg140 produces β-kallikrein in some experiments. FXI is a 160-kDa dimer of two 80-kDa polypeptides (1 shown). It is converted to FXIa by cleavage after Arg369. The noncatalytic portions of PK and FXI contain 4 apple domains, designated A1 to A4 from the N-terminus. FXII is an 80-kDa polypeptide. Cleavage after Arg353 is responsible for formation of the protease αFXIIa. The noncatalytic portion of FXII contains fibronectin type 2 (F2), epidermal growth factor (EGF), fibronectin type 1 (F1), and kringle (K) domains, and a proline–rich region (PRR). (B) Nonreducing SDS-PAGE of ∼2-μg samples of purified PK or FXI. Left panel, Plasma PK (pPK), recombinant wild-type PK (WT), and PK variants with alanine replacing Arg371 (PK-R371A or R371A) or Ser559 (PK-S559A or S559A). Right panel, Plasma FXI (pFXI), recombinant wild-type FXI (WT), and FXI variants FXI-R369A (R369A) or FXI-S557A (S557A). (C) Plasma PK, PK-WT, PK-R371A, or PK-S559A, 200 nM, were incubated with (+) or without (−) 40 nM FXIIa at 37°C. After 10 minutes, reactions were stopped with reducing sample buffer, size fractionated by SDS-PAGE, and analyzed by western blot using polyclonal anti-PK IgG. For panel B, positions of molecular mass standards in kilodaltons are shown to the left of the images. For panel C, positions of standards for PK, the heavy chain (HC) and light chain (LC) of PKa, and a fragment representing part of β-kallikrein are shown at the right of the figure. (D) Cleavages of S-2302 by PK-WT, PK-R371A, or PK-S559A after digestion with FXIIa as described in panel C (FXIIa was blocked with 1 μM CTI). (E) Clotting times in an aPTT assay for pooled normal plasma (PNP), PK-deficient plasma (C) or PK-deficient plasma supplemented with plasma-derived PK or with recombinant PK-WT (WT), PK-R371A (371), or PK-S559A (559). Each symbol indicates 1 clotting time and the horizontal bars indicate means for each group ± 1 standard deviation. *P ≤ .05 compare with pooled normal plasma. HC, heavy chain; LC, light chain.
Recombinant proteins. (A) Schematic diagrams of contact activation proteases. Catalytic domains are indicated by gray boxes, and noncatalytic domains by white boxes. Positions of active site serine residues are indicated by black bars. Sites of proteolysis during activation are indicated by arrows, with black arrows indicating sites of cleavage required for full protease activity. PK is an 86-kDa polypeptide that is cleaved after Arg371 to form α-kallikrein (PKa). A second cleavage after Arg140 produces β-kallikrein in some experiments. FXI is a 160-kDa dimer of two 80-kDa polypeptides (1 shown). It is converted to FXIa by cleavage after Arg369. The noncatalytic portions of PK and FXI contain 4 apple domains, designated A1 to A4 from the N-terminus. FXII is an 80-kDa polypeptide. Cleavage after Arg353 is responsible for formation of the protease αFXIIa. The noncatalytic portion of FXII contains fibronectin type 2 (F2), epidermal growth factor (EGF), fibronectin type 1 (F1), and kringle (K) domains, and a proline–rich region (PRR). (B) Nonreducing SDS-PAGE of ∼2-μg samples of purified PK or FXI. Left panel, Plasma PK (pPK), recombinant wild-type PK (WT), and PK variants with alanine replacing Arg371 (PK-R371A or R371A) or Ser559 (PK-S559A or S559A). Right panel, Plasma FXI (pFXI), recombinant wild-type FXI (WT), and FXI variants FXI-R369A (R369A) or FXI-S557A (S557A). (C) Plasma PK, PK-WT, PK-R371A, or PK-S559A, 200 nM, were incubated with (+) or without (−) 40 nM FXIIa at 37°C. After 10 minutes, reactions were stopped with reducing sample buffer, size fractionated by SDS-PAGE, and analyzed by western blot using polyclonal anti-PK IgG. For panel B, positions of molecular mass standards in kilodaltons are shown to the left of the images. For panel C, positions of standards for PK, the heavy chain (HC) and light chain (LC) of PKa, and a fragment representing part of β-kallikrein are shown at the right of the figure. (D) Cleavages of S-2302 by PK-WT, PK-R371A, or PK-S559A after digestion with FXIIa as described in panel C (FXIIa was blocked with 1 μM CTI). (E) Clotting times in an aPTT assay for pooled normal plasma (PNP), PK-deficient plasma (C) or PK-deficient plasma supplemented with plasma-derived PK or with recombinant PK-WT (WT), PK-R371A (371), or PK-S559A (559). Each symbol indicates 1 clotting time and the horizontal bars indicate means for each group ± 1 standard deviation. *P ≤ .05 compare with pooled normal plasma. HC, heavy chain; LC, light chain.
Protein purification
FXII and PK were purified by anion-exchange chromatography.17,18 Preparation of δFXII-3C, a truncated FXII (amino acids 308-596) lacking most of the heavy chain was described.17 Proteins were stored in 4 mM sodium acetate, 150 mM NaCl, pH 5.2 at −80°C. FXI was purified by sequential SP-Sepharose and heparin-agarose chromatography, and stored in 4 mM Tris-HCl, 150 mM NaCl, pH 7.4 at −80°C.
Western blots
Reactions were conducted in polypropylene tubes coated with PEG-20 000. HK (200 nM) with or without PK (200 nM) was incubated in phosphate-buffered saline (PBS; 10 mM phosphate, pH 7.4, 137 mM NaCl, 2.7 mM KCl) at 37°C. Some reactions contained 750 nM CTI or C1-INH. FXII (200 nM) with or without PK (200 nM) or FXI (30 nM) was incubated in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% PEG-8000, 10 μM ZnCl2) at 37°C. Some reactions contained 70 μM poly-P, 25 μg/mL genomic DNA, 1 μg/mL dextran sulfate (DXS), or 15% PTT-A reagent. At various times, aliquots were removed into sodium dodecyl sulfate (SDS)-sample buffer, size fractionated by SDS–polyacrylamide gel electrophoresis (PAGE) (12% acrylamide), and transferred to nitrocellulose membranes. Blots of reduced HK or PK were probed with polyclonal IgG to those proteins. Nonreducing blots of FXII/FXIIa were probed with IgG D06, which preferentially recognizes FXIIa. For all blots, detection was performed with an HRP-conjugated secondary antibody and chemiluminescence.
Chromogenic assays
Continuous assays
Assays were conducted in 96-well plates coated with PEG-20 000. Activation of FXII (FXII-WT or δFXII-3C, 50 nM) and/or PK (50 nM) was monitored in 100 μL of HEPES buffer at 37°C. Reactions contained 200 μM S-2302 and changes in optical density (OD) 405 nm were monitored on a microplate reader.
Discontinuous assay
Activation of FXII (200 nM) by PK and variants (200 nM) in the presence of poly-P (70 μM) were performed in HEPES buffer at 37°C in the presence or absence HK (200 nM). Reactions were stopped after 60 minutes with polybrene (0.1 mg/mL) and cleavage of 500 μM S-2302 (ΔOD 405 nm) was monitored.
Kinetic analysis of HK cleavage
HK (12-1600 nM) was incubated with PKa (1 nM) or PK-R371A (200 nM) in PBS for 2 hours at 37°C. Reactions were stopped by adding 9 volumes of ice-cold ethanol and immediately freezing at −80°C. Thawed samples underwent centrifugation at 10 000g for 1 hour at 4°C. The BK-containing supernatant was evaporated, and the remaining nonvolatile material was reconstituted with an enzyme-linked immunosorbent assay (ELISA) buffer. BK was measured by ELISA (ELISA BK; Enzo Life Sciences). Results were compared with a standard curve prepared with pure BK. Km and kcat were determined by nonlinear least-squares fitting using Scientist software (Micromath).
Kinetic analysis of FXII activation
FXII-S544A (3-1600 nM) was incubated with PKa (1 nM) or PK-R371A (200 nM) in the presence of poly-P (70 μM) in HEPES buffer at 37°C for 2 hours. Reactions were stopped by adding polybrene to 0.1 mg/mL and anti-PKa IgG HO3 to 5 nM. FXIIa was measured by ELISA using the anti-FXIIa IgG DO6 as the capture antibody, and an HRP-conjugated anti-FXII IgG for detection. FXIIa generated in each reaction was calculated from a standard curve constructed with pure FXIIa (supplemental Figure 1, available on the Blood Web site). Km and kcat were determined by nonlinear least-squares fitting using Scientist software (Micromath).
Plasma-clotting assays
PK-deficient plasma (30 μL) was mixed with 30 μL of PK (600 nM) in Tris-buffered saline (20 mM Tris-HCl, 136 mM NaCl, pH 7.4). PTT-A reagent (30 μL) was added followed by incubation for 5 minutes at 37°C. CaCl2 (30 μL of 25 mM solution) was added, and time to clot formation was measured on an ST-4 Coagulation Analyzer (Diagnostica Stago). Results were compared with healthy or PK-deficient plasma mixed with an equal volume of Tris-buffered saline.
Thrombin generation
Thrombin generation was measured in 96-well polypropylene plates on a Fluoroskan Ascent analyzer.30,31 Briefly, PK-deficient plasma was supplemented with PK (600 nM), 415 mM Z-Gly-Gly-Arg-AMC, and vehicle or PTT-A reagent (13% final volume). The supplemented plasma (40 μL) was mixed with 20 mM HEPES, pH 7.4, 100 mM CaCl2, 6% bovine serum albumin (10 μL), and changes in fluorescence (excitation, λ 390 nm; emission, λ 460 nm) were monitored. Peak thrombin generation, time to peak thrombin generation, and area under the curve (endogenous thrombin potential [ETP]) were calculated using Thrombinoscope Analysis software (version 3.0).
Carotid artery thrombosis model
Procedures with mice were approved by the Vanderbilt University Animal Care and Use Committee. PK-deficient C57Bl/6 mice32 were anesthetized with 50 mg/kg intraperitoneal pentobarbital. The right common carotid artery was exposed and fitted with a Doppler probe (model 0.5 VB; Transonic System). PK (50 μg in 200 μL of PBS) was infused through the internal jugular vein. Five minutes later, two 1 × 1.5-mm filter papers (Schleicher & Schuell) saturated with 5% FeCl3 were applied to opposite sides of the artery for 3 minutes. Flow was monitored for 30 minutes.
Statistical analysis
Statistics were performed using GraphPad Prism 5 (La Jolla, CA). Data are expressed as means plus or minus standard deviation. Statistical significance for data in Figures 1E and 4C was determined by using the Mann-Whitney U test. P ≤ .05 was considered statistically significant.
Results
Recombinant PK characteristics
Purified recombinant PKs are shown in Figure 1B. FXIIa cleaves plasma-derived PK, PK-WT, and PK-S559A similarly (Figure 1C). PK-R371A lacks the conserved arginine at the activation site and is not cleaved (Figure 1C), confirming its resistance to conversion to PKa. Of the PK species incubated with FXIIa, only PK-WT demonstrates amidolytic activity with the tripeptide S-2302 (Figure 1D). This is expected, as PK-R371A is not converted to PKa, and PKa-S559A lacks the active site serine required for catalytic activity. In aPTT assays, plasma PK and PK-WT, but not PK-R371A or PK-S559A, shorten the clotting time of PK-deficient plasma (Figure 1E).
Single-chain PK cleavage of HK
PKa cleaves HK after Lys380 and Arg389, releasing BK (Arg381-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg389),3,4 and converting the 110-kDa HK band on SDS-PAGE to the 65-kDa and 45-kDa bands representing cleaved HK (HKa) (Figure 2A; supplemental Figure 2). PK-WT also catalyzes HK conversion to HKa by a process inhibited by C1-INH (Figure 2B second column). In the study by Joseph et al, plasma-derived PK cleavage of HK was inhibited by CTI.26 We initially obtained similar results (supplemental Figure 3A). CTI is a known FXIIa inhibitor,30,33,34 but does not inhibit PKa.35 We were concerned that the apparent inhibition of PK by CTI actually reflected contamination of the system with FXII or FXIIa that could activate PK. We identified FXII as a minor contaminant of HK preparations (supplemental Figure 3B). The effect of FXII on the system was verified by observing that the anti-FXIIa IgG D06 reduced HK cleavage by PK (supplemental Figure 3C). When HK lacking obvious FXII contamination was used (Figure 2B second column), the effect of CTI was lost.

HK cleavage by PKa and PK. Western blots of time courses of cleavage of HK (200 nM) by (A) PKa (2 nM) or (B) recombinant PK species (200 nM) in PBS at 37°C. In panel B, “vehicle” indicates reactions without PK. Some reactions in panel B were run in the presence of 750 nM C1INH or 750 nM CTI. At indicated time points, samples were removed into reducing sample buffer, size fractionated by SDS-PAGE, and analyzed by western blotting using polyclonal anti-HK IgG. Positions of standards for uncleaved HK (HK) and heavy and light chains of cleaved HK (HKa) are indicated at the right of each image. (C) Varying concentrations of HK were incubated with 1 nM α-kallikrein (left panel) or 200 nM PK-R371A (right panel) in PBS at 37°C. After 2 hours of incubation, reactions were stopped by ice-cold ethanol and BK concentration was determined by ELISA. Each point represents a single measurement. (D) Kinetic parameters for cleavage of HK by PKa or PK-R371A determined from the curves in panel C.
HK cleavage by PKa and PK. Western blots of time courses of cleavage of HK (200 nM) by (A) PKa (2 nM) or (B) recombinant PK species (200 nM) in PBS at 37°C. In panel B, “vehicle” indicates reactions without PK. Some reactions in panel B were run in the presence of 750 nM C1INH or 750 nM CTI. At indicated time points, samples were removed into reducing sample buffer, size fractionated by SDS-PAGE, and analyzed by western blotting using polyclonal anti-HK IgG. Positions of standards for uncleaved HK (HK) and heavy and light chains of cleaved HK (HKa) are indicated at the right of each image. (C) Varying concentrations of HK were incubated with 1 nM α-kallikrein (left panel) or 200 nM PK-R371A (right panel) in PBS at 37°C. After 2 hours of incubation, reactions were stopped by ice-cold ethanol and BK concentration was determined by ELISA. Each point represents a single measurement. (D) Kinetic parameters for cleavage of HK by PKa or PK-R371A determined from the curves in panel C.
Although we felt that we could limit FXII/FXIIa activation of PK-WT by screening HK preparations for FXII, we could not rule out that traces of PKa were present in PK-WT preparations, or were generated after initiation of reactions. We addressed this by replacing PK-WT with PK-R371A (single-chain PK), a protein that is not converted to PKa. PK-R371A catalyzed HK cleavage similarly to PK-WT (Figure 2B third column). The reaction was inhibited by C1-INH (Figure 2B) and by an IgG to the PKa active site (H03), but not by CTI (Figure 2B third column) or anti-FXIIa IgG D06 (supplemental Figure 3D). PK activity required an intact catalytic triad, as PK lacking an active site serine residue (PK-S559A) did not cleave HK (Figure 2B fourth column). Orthophosphate polymers (poly-P) are found in many tissues.11,35-37 Platelets release poly-P species of 60 to 100 phosphate units upon activation.36 poly-P accelerates FXII-PK reciprocal activation, promoting contact activation.16-18,38 Although poly-P modestly enhanced HK cleavage by PKa, it had no appreciable effect on HK cleavage by PK-R371A (supplemental Figure 4).
Kinetics of single-chain PK cleavage of HK
PKa cleaved HK to release BK with Km of 440 ± 100 nM and kcat of 0.46 ± 0.05/min (Figure 2C left panel). Parameters for PK-R371A cleavage of HK (Km, 139 ± 30 nM; kcat, 9.7 ± 0.6 × 10−5/min) (Figure 2C right panel) indicate a catalytic efficiency (kcat/Km, 1.06 × 10−3 nM−1min−1 vs 6.95 × 10−7 nM−1min−1) ∼1500-fold lower than for PKa (Figure 2D). Plasma-derived PK cleavage of HK was reported to be stoichiometric.26 The reaction we observed, even with 200 nM PK-R371A, does not exclude this possibility as, at saturating HK concentrations, only ∼2.4 nM BK was generated (∼20 pM BK/min) after 2 hours. However, at 200 nM HK, the rates did not reach an experimental plateau value, consistent with Km of 139 nM, or ∼60% saturation of PK-R371A. The earlier report showed complete HK cleavage at limiting HK concentrations,26 which is not proof of stoichiometry. However, release of equivalent BK at limiting PK concentrations and saturating HK was observed, suggesting 1 turnover per cycle, consistent with HKa binding to PK that prevents further HK proteolysis.
Single-chain PK cleavage of FXII
PKa catalyzes FXII activation by a reaction that is accelerated by negatively charged surfaces and HK.17,18,38 When examining FXII activation by PKa in the presence of a surface, FXII autoactivation complicates interpretation. To remove this effect, the substrate in the following studies, FXII-S544A, lacks an active site serine and is catalytically inactive.17 FXII-S544A is cleaved by PKa after Arg353similarly to FXII-WT (Figure 1C). The FXIIa conformation adopted by FXII-S544A is detected on western blots using the FXIIa-specific IgG D06 (Figure 3A).17
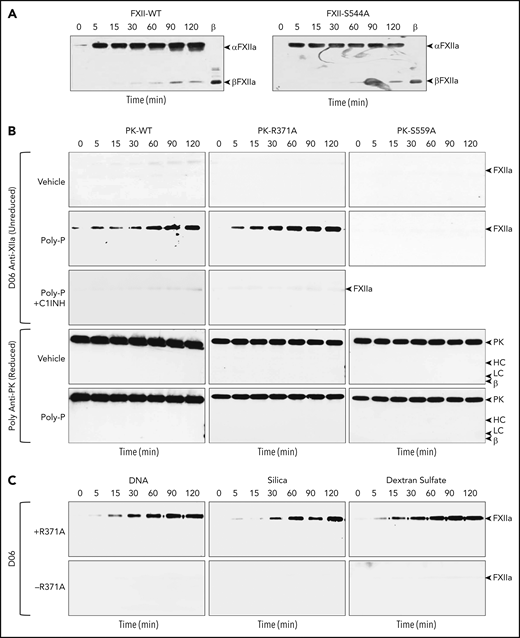
FXII cleavage by PKa and PK. Western blots of time courses of cleavage of FXII or FXII-S544A (200 nM) by (A) PKa (50 nM). At indicated time points, samples were removed into nonreducing sample buffer, size fractionated by SDS-PAGE, and analyzed by western blotting using monoclonal anti-FXIIa IgG D06. Positions of standards for αFXIIa and βFXIIa are indicated at the right of each image. (B) Western blots of time courses of cleavage of FXII-S544A (200 nM) by recombinant PK (PK-WT, PK-R371A, or PK-S544A) in the presence of vehicle (top row), poly-P (70 μM, second row), or poly-P and C1INH (750 nM, third row). Blots were processed as in panel A. Positions of a standard for αFXIIa are indicated at the right of each image. The fourth and fifth rows are western blots for PK, using the same samples for reactions with vehicle and poly-P used in the first 3 rows. Positions of standards for PK, the heavy (HC) and light (LC) chains of PKa, and a heavy-chain fragment from β-kallikrein are indicated at the right of each image. (C) Western blots of time courses of cleavage of FXII-S544A (200 nM) in the presence (+) or absence (−) of PK-R371A, with human genomic DNA (25 μg/mL), 15% (vol/vol) silica containing PTT-A reagent (silica), or DXS (1 μg/mL). Blots were processed as in panel A. Positions of a standard for αFXIIa are indicated at the right of each image.
FXII cleavage by PKa and PK. Western blots of time courses of cleavage of FXII or FXII-S544A (200 nM) by (A) PKa (50 nM). At indicated time points, samples were removed into nonreducing sample buffer, size fractionated by SDS-PAGE, and analyzed by western blotting using monoclonal anti-FXIIa IgG D06. Positions of standards for αFXIIa and βFXIIa are indicated at the right of each image. (B) Western blots of time courses of cleavage of FXII-S544A (200 nM) by recombinant PK (PK-WT, PK-R371A, or PK-S544A) in the presence of vehicle (top row), poly-P (70 μM, second row), or poly-P and C1INH (750 nM, third row). Blots were processed as in panel A. Positions of a standard for αFXIIa are indicated at the right of each image. The fourth and fifth rows are western blots for PK, using the same samples for reactions with vehicle and poly-P used in the first 3 rows. Positions of standards for PK, the heavy (HC) and light (LC) chains of PKa, and a heavy-chain fragment from β-kallikrein are indicated at the right of each image. (C) Western blots of time courses of cleavage of FXII-S544A (200 nM) in the presence (+) or absence (−) of PK-R371A, with human genomic DNA (25 μg/mL), 15% (vol/vol) silica containing PTT-A reagent (silica), or DXS (1 μg/mL). Blots were processed as in panel A. Positions of a standard for αFXIIa are indicated at the right of each image.
Neither PK-WT nor PK-R371A cleaved FXII-S544A in the absence of a surface (Figure 3B top row). In the presence of poly-P, however, PK-WT and PK-R371A, but not PK-S559A, converted FXII-S544A to FXIIa-S544A (Figure 3B second row). The reactions are inhibited by C1-INH (Figure 3B third row), and, during the reactions, PK appears to remain in its single-chain form (Figure 3B bottom two rows). DNA, silica, and DXS all induce contact activation in plasma, and support PK-R371A–catalyzed conversion of FXII-S544A to FXIIa-S544A (Figure 3C).
Kinetics of single-chain PK cleavage of FXII
PKa cleaves FXII in the presence of poly-P with Km of 117 ± 22 nM and kcat of 1.18 ± 0.09/min, whereas PK-R371A cleaves FXII with Km of 1781 ± 500 nM and kcat of 8.0 ± 1.0 × 10−3/min (Figure 4A-B). The catalytic efficiency (kcat/Km) of PK-R371A cleavage of FXII is ∼23 000-fold lower than for cleavage by PKa (4.32 × 10−7 nM−1min−1 vs 1.01 × 10−2 nM−1min−1; Figure 4B). HK is a cofactor for PKa activation of FXII.3,4,10,38,39 Adding HK to reactions with FXII, PK-R371A and poly-P increases FXIIa generation approximately eightfold, but had no effect in the absence of poly-P (Figure 4C). In Figure 4C, FXIIa activity in reactions with poly-P in the absence of PK or with PK-S559A reflects FXII autoactivation.
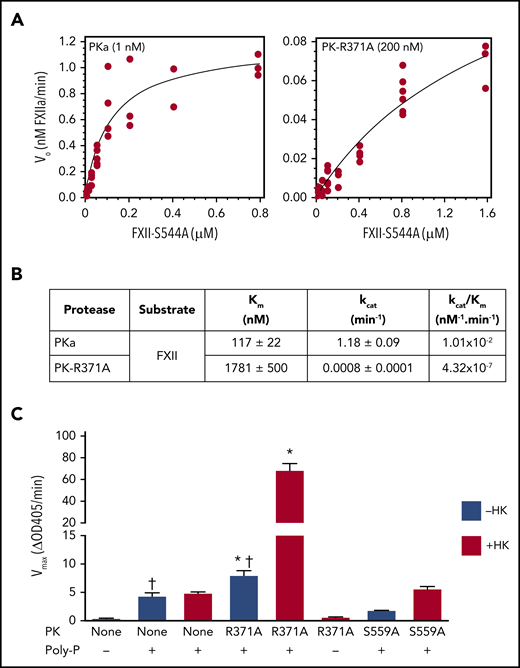
Kinetics of FXII cleavage by PK and PKa. (A) Varying concentrations of FXII-S544A were incubated with 1 nM PKa (left panel) or 200 nM PK-R371A (right panel) in HEPES buffer at 37°C in the presence of 70 μM poly-P. After 120 minutes, reactions were stopped by adding polybrene (0.1 mg/mL) and the antikallikrein IgG HO3 (5 nM). Conversion of FXII-S544A to the FXIIa conformation was measured by ELISA. (B) Kinetic parameters of FXII-S544A cleavage by PKa or PK-R371A in the presence of poly-P determined from the curves in panel A. (C) FXII-WT (200 nM) was incubated in the absence (−) or presence (+) of 70 μM poly-P. Reactions were run in the absence of PK (None) or in the presence of 200 nM PK-R371A or PK-S559A. Blue columns are reactions run in the absence of HK; red columns indicate reactions run in the presence of 200 nM HK. Reactions were stopped after 60 minutes with polybrene (0.1 mg/mL), and FXIIa cleavage of S-2302 (500 μM) was measured at OD 405 nm. *Compares FXII activation by PK-R371A in the presence or absence of HK (P = .0001); †compares FXII activation in the presence or absence of PK-R371A (P = .01).
Kinetics of FXII cleavage by PK and PKa. (A) Varying concentrations of FXII-S544A were incubated with 1 nM PKa (left panel) or 200 nM PK-R371A (right panel) in HEPES buffer at 37°C in the presence of 70 μM poly-P. After 120 minutes, reactions were stopped by adding polybrene (0.1 mg/mL) and the antikallikrein IgG HO3 (5 nM). Conversion of FXII-S544A to the FXIIa conformation was measured by ELISA. (B) Kinetic parameters of FXII-S544A cleavage by PKa or PK-R371A in the presence of poly-P determined from the curves in panel A. (C) FXII-WT (200 nM) was incubated in the absence (−) or presence (+) of 70 μM poly-P. Reactions were run in the absence of PK (None) or in the presence of 200 nM PK-R371A or PK-S559A. Blue columns are reactions run in the absence of HK; red columns indicate reactions run in the presence of 200 nM HK. Reactions were stopped after 60 minutes with polybrene (0.1 mg/mL), and FXIIa cleavage of S-2302 (500 μM) was measured at OD 405 nm. *Compares FXII activation by PK-R371A in the presence or absence of HK (P = .0001); †compares FXII activation in the presence or absence of PK-R371A (P = .01).
Recently, we described the properties of δFXII, a truncated FXII lacking the heavy-chain N-terminal to residue 309 that is generated in some patients with hereditary angioedema.18 δFXII is activated by PKa in the absence of a surface with ∼15-fold higher catalytic efficiency than FXII-WT. The reaction is not enhanced by surfaces because δFXII lacks a surface-binding heavy chain. Although δFXII readily participates in reciprocal activation with PK-WT, it is not activated appreciably by PK-R371A (supplemental Figure 5), indicating that FXII activation by single-chain PK requires FXII to be surface-bound.
Single-chain PK activity in plasma
PK-WT and plasma-derived PK support thrombin generation comparably in plasma in which contact activation is induced with a silica-based reagent (Figure 5A). The proteins have little effect without silica (Figure 5B). In reactions with silica, there is some PK-independent thrombin generation (reactions with vehicle or PK-S559A) due to FXII autoactivation (Figure 5A). Although PK-R371A has insufficient activity to affect the aPTT-clotting time (Figure 1E), some activity is observed in the more sensitive thrombin-generation assay. Compared with PK-WT (Figure 5C), PK-R371A exhibited lower ETP (3235 ± 18 nM.min vs 2588 ± 23 nM.min, respectively) and peak thrombin generation (846 ± 8 nM vs 392 ± 8 nM), and a longer time to peak generation (7.8 ± 0.3 minutes vs 20.1 ± 0.2 minutes).
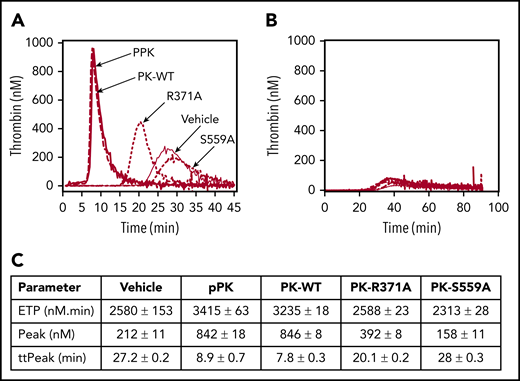
Effects of PK on thrombin generation in plasma. (A) Thrombin generation in PK-deficient plasma induced to clot with PTT-A reagent. Reactions were run in the absence of PK (vehicle; solid gray line) or in the presence of 600 nM plasma PK (pPK; heavy solid line), PK-WT (dashed line), PK-R371A (dotted line), or PK-S559A (dash-dotted line). (B) Same as panel A, but no PTT-A reagent was added. (C) Thrombin-generation parameters derived from the curves in panel A. Each curve represents means for reactions run in triplicate.
Effects of PK on thrombin generation in plasma. (A) Thrombin generation in PK-deficient plasma induced to clot with PTT-A reagent. Reactions were run in the absence of PK (vehicle; solid gray line) or in the presence of 600 nM plasma PK (pPK; heavy solid line), PK-WT (dashed line), PK-R371A (dotted line), or PK-S559A (dash-dotted line). (B) Same as panel A, but no PTT-A reagent was added. (C) Thrombin-generation parameters derived from the curves in panel A. Each curve represents means for reactions run in triplicate.
Single-chain PK in thrombosis models
PK-deficient (Klkb1−/−) mice are protected from carotid artery thrombosis induced by applying FeCl3 to the vessel exterior (supplemental Figure 6).40 Infusing PK-WT restored thrombus formation in Klkb1−/− mice, whereas PK-R371A did not. These data are consistent with those from the aPTT and thrombin-generation assays, and indicate that, whereas PK-R371A expresses a low level of enzymatic activity, PKa is required to generate sufficient thrombin to support clot formation.
Single-chain FXI (FXI-R369A)
FXI is a homolog of PK.29,41,42 Like PK, FXI is a FXIIa substrate, and like PKa, activated FXI (FXIa) converts FXII to FXIIa in a reaction accelerated by poly-P.43 We prepared FXI that cannot be converted to FXIa (FXI-R369A, single-chain FXI; supplemental Figure 7A) and tested its ability to cleave FXII-S544A. Plasma-derived FXIa converted FXII to FXIIa in a reaction accelerated by poly-P (supplemental Figure 7B) that is so rapid that FXIIa undergoes degradation to βFXIIa and other forms over the course of the assay. In reactions with FXI-WT, some FXIIa-S544A is observed at late time points independently of poly-P, possibly reflecting contamination with FXIa. FXI-R369A does not convert FXII-S544A to FXIIa (supplemental Figure 7B bottom row). Similarly, FXI-R369A did not activate factor IX (the major substrate of FXIa; supplemental Figure 7C), nor did it catalyze FXI cleavage in the presence of poly-P (autoactivation; supplemental Figure 7D). These findings suggest that single-chain FXI does not express proteolytic activity toward FXIa substrates.
Discussion
In 1978, Huber and Bode proposed a mechanism for activating trypsin-like proteases in which an inactive zymogen (usually a single polypeptide) is irreversibly converted to an active enzyme by cleavage after an internal arginine (typically Arg15 using chymotrypsin numbering).44 The new N terminus of the catalytic domain (residue 16) created by the cleavage forms a hydrogen bond with Asp194, which supports protease active site formation.44,45 This iconic model explains onset of enzymatic activity and accounts for the behavior of protease cascades, such as those involved in blood coagulation and complement activation. However, the mechanism is not easily reconciled with certain processes such as protease autoactivation. Furthermore, when considering a sequential cascade of proteolytic reactions, it does not account for the activity that initiates the sequence.
Recently, Chakraborty and coworkers proposed an alternative interpretation for activation and activity of trypsin-like proteases based on work with the zymogen prothrombin and its protease form thrombin.46,47 They suggest that a protease exists as a dynamic ensemble of conformations with the active sites either accessible (E) or inaccessible (E*) to a substrate. Typically, the catalytic apparatus of the precursor is largely, but not necessarily entirely, in the E* form. Cleavage after Arg15 results in a shift to species where the E form predominates, corresponding to full enzymatic activity. Depending on the protease, the putative zymogen may exhibit measurable activity if a sufficient fraction adopts the E form either before or after engaging a substrate. As examples, the fibrinolytic proteases tissue plasminogen activator and urokinase display substantial activity in their single-chain forms as a consequence of structural features that maintain the active site in an open (E) conformation.48,49 These findings seem relevant to the observed behaviors of the contact protease precursors PK and FXII.
Mixing PK and FXII in the absence of a surface results in their reciprocal conversion to PKa and FXIIa.16-18 It is our observation that pretreatment with inhibitors to neutralize contaminating PKa and FXIIa has little impact on this process. Revenko et al showed that reducing PK in mice with an antisense oligonucleotide leads to an increase in plasma FXII (presumably because it is not consumed) and reduced FXIIa formation, whereas reducing FXII leads to increased plasma PK and reduced PKa.7 This suggests that reciprocal FXII-PK activation occurs at a basal level in vivo. The nature of the activity that initiates (and perhaps sustains) the process is debated, and proteolytic and nonproteolytic mechanisms have been proposed.23,24,50-53 We showed that FXII restricted to its single-chain form catalyzes PK activation.17 The FXII active site must, therefore, be able to adopt an open conformation independently of cleavage after Arg353 (homologous to chymotrypsin Arg15) and formation of the Arg15-Asp194 hydrogen bond.
We propose that PK in its single-chain form, like FXII, expresses proteolytic activity. Previously, Joseph and colleagues reported that plasma-derived PK cleaves HK in a stoichiometric interaction inhibited by C1-INH or a peptide that blocks HK binding to PK.26,27 In the current study, we take into account the possibility that traces of PKa or FXIIa in preparations of plasma-derived proteins were responsible for apparent PK activity by testing PK-R371A, a single-chain species that cannot be converted to PKa. We show that PK-R371A cleaves HK in the absence of a surface and activates FXII in the presence of a surface. That these reactions are inhibited by C1-INH and an antibody designed to bind to the PKa active site supports the notion that the catalytic apparatus of some protease precursors adopt open conformations that permit proteolysis of peptide bonds.
Single-chain forms of PK and FXII exhibit very low amidolytic activity when small tripeptides are used as substrates. This raises the possibility that their catalytic domains are mostly in a closed conformation until a macromolecular substrate is bound.17,54 The substrate may bind one or more exosites remote from the active site on the catalytic domain or noncatalytic parts of the protein. This, in turn, may allosterically promote formation of an open active site (induced fit).55 For PK, the apple domains (Figure 1A) form a platform, with the catalytic domain resting on one face of the platform, and a binding site for HK resting on the other face.56,57 Joseph et al noted that interfering with HK binding to PK with a peptide reduces the capacity of plasma-derived PK to cleave HK.26 Interestingly, the inhibitory peptide did not interfere with PKa cleavage of HK, suggesting that conversion of PK to PKa unmasks a second HK-binding exosite. Recent studies by Li et al56 and Bar Barroeta et al58 indicate that activation of PK or its homolog FXI involves a 180° pivoting of the catalytic domain on the apple domain disk, exposing areas of the surface of apple domain 3, where substrate-binding sites may reside.59
The catalytic efficiency of HK cleavage by single-chain PK is ∼1500 times lower than for PKa, mirroring the low activity of single-chain FXII.17 This suggests that PKa is required for substantial increases in BK generation, and is consistent with the impression that BK generation during bouts of angioedema in hereditary angioedema patients8-10 or in sepsis60,61 involves PK conversion to PKa. However, the situation may be different for basal BK production in normal health.7 Here, the rate of PKa generation appears to be relatively low,62 whereas the ratio of PK to PKa is probably high. Given the PK plasma concentration (500-600 nM), it is conceivable that PK cleavage of HK contributes significantly to basal HK cleavage and BK formation.
In contrast to our experience with PK activation by single-chain FXII,17 single-chain PK did not catalyze FXII activation without a surface. This suggests that the initial step in surface-independent reciprocal activation is FXII-catalyzed conversion of PK to PKa.18 In the presence of poly-P, however, single-chain PK catalyzes FXII activation. The reaction is several orders of magnitude weaker that with PKa, consistent with the impression that PKa is required to form physiologic amounts of FXIIa, and with our observations that PK-R371A has a negligible effect in an aPTT assay and a mouse thrombosis model. Like surface-dependent FXII activation by single-chain FXII, then, FXII activation by single-chain PK is most likely relevant for initiating contact activation when blood is first exposed to a surface and levels of PKa and FXIIa are very low. Finally, in the absence of a surface, PKa is a weak FXII activator, and FXII does not autoactivate. These observations, along with data for the single-chain forms of PK and FXII, suggest that surface binding causes conformational changes in FXII that facilitate cleavage after Arg353 by PK, FXII, and their activated forms. This is consistent with work indicating that surface binding changes the orientation of the FXII heavy chain relative to the activation cleavage site.15,18,63
The activities inherent in single-chain PK and FXII provide a solution to the chicken-or-the-egg dilemma regarding initiation of reciprocal FXII-PK activation and contact activation. The findings imply that using active site inhibitors of PKa or FXIIa to treat angioedema or thrombosis could provide additional benefit by retarding PK and FXII reciprocal activation through inhibition of the activity inherent in these precursor proteins.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
This work was supported by awards HL81326, HL58837, and HL140025 (D.G.) and HL130018 (I.M.V.) from the National Institutes of Health National Heart, Lung, and Blood Institute.
Authorship
Contribution: I.I. conducted experiments on PK activity in purified systems and in plasma, and contributed to the writing of the manuscript; A.M. designed and conducted thrombin-generation studies and contributed to the writing of the manuscript; Q.C. and B.M. performed studies on mice; M.-f.S. and S.K.D. prepared and characterized recombinant proteins and antibodies; I.M.V. conducted kinetic analyses; and K.J., A.P.K., and D.G. contributed to study design and data interpretation and to the writing of the manuscript.
Conflict-of-interest disclosure: D.G. is a consultant for several pharmaceutical companies with interests in targeting components of contact activation for therapeutic purposes. The remaining authors declare no competing financial interests.
Correspondence: David Gailani, Vanderbilt University Medical Center, Room 4918, The Vanderbilt Clinic, 1301 Medical Center Dr, Nashville, TN 37232; e-mail: dave.gailani@vanderbilt.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal