

Key Points
The N terminus of ERFE binds BMP6 with nanomolar affinity and is sufficient to inhibit BMP signaling and suppress hepcidin in vivo.
Anti–N-terminal ERFE antibodies prevent EPO-induced hepcidin suppression and decrease iron accumulation and anemia in thalassemic mice.

Abstract
Erythroferrone (ERFE) is produced by erythroblasts in response to erythropoietin (EPO) and acts in the liver to prevent hepcidin stimulation by BMP6. Hepcidin suppression allows for the mobilization of iron to the bone marrow for the production of red blood cells. Aberrantly high circulating ERFE in conditions of stress erythropoiesis, such as in patients with β-thalassemia, promotes the tissue iron accumulation that substantially contributes to morbidity in these patients. Here we developed antibodies against ERFE to prevent hepcidin suppression and to correct the iron loading phenotype in a mouse model of β-thalassemia [Hbb(th3/+) mice] and used these antibodies as tools to further characterize ERFE’s mechanism of action. We show that ERFE binds to BMP6 with nanomolar affinity and binds BMP2 and BMP4 with somewhat weaker affinities. We found that BMP6 binds the N-terminal domain of ERFE, and a polypeptide derived from the N terminus of ERFE was sufficient to cause hepcidin suppression in Huh7 hepatoma cells and in wild-type mice. Anti-ERFE antibodies targeting the N-terminal domain prevented hepcidin suppression in ERFE-treated Huh7 cells and in EPO-treated mice. Finally, we observed a decrease in splenomegaly and serum and liver iron in anti–ERFE-treated Hbb(th3/+) mice, accompanied by an increase in red blood cells and hemoglobin and a decrease in reticulocyte counts. In summary, we show that ERFE binds BMP6 directly and with high affinity, and that antibodies targeting the N-terminal domain of ERFE that prevent ERFE–BMP6 interactions constitute a potential therapeutic tool for iron loading anemias.
Introduction
β-thalassemia is an inherited hemoglobinopathy characterized by dysfunction or deletion of the β globin genes, leading to hemolytic anemia, ineffective erythropoiesis, and iron overload.1,2 Approximately 1.5% of the population worldwide are carriers of β-thalassemia3 ; homozygous or compound heterozygous states result in thalassemia intermedia or major (the latter requiring regular blood transfusions).4 A main cause of morbidity in these patients is iron overload, which accumulates in several tissues, especially the liver. It causes damage due to iron toxicity5 and is associated with hepatic fibrosis6 and hepatocellular carcinoma,7,8 cardiac failure and arrhythmia, endocrine failure (ie, hypogonadism, diabetes), and osteoporosis9 may also occur. Current treatments can have undesirable side effects: regular blood transfusions (in thalassemia major) significantly worsen iron accumulation; iron chelators alleviate iron loading but may require intravenous or subcutaneous administration (although oral administration is also currently used), and they may cause gastrointestinal disturbances and/or kidney damage.10 Understanding the mechanism underlying iron accumulation may contribute to the design of better therapies to improve the clinical outcome.
Enhanced erythropoiesis requires augmented iron availability for heme production.11 This is achieved by suppression of hepcidin, a hepatic hormone that regulates iron absorption and distribution by inhibiting the iron exporter ferroportin.12-14 Hepcidin expression is modulated by the BMP/SMAD signaling pathway: binding of bone morphogenetic proteins (BMPs) to BMP receptors in the membrane of hepatocytes causes phosphorylation of cytosolic SMADs (SMAD1/5/8) that translocate to the nucleus complexed with SMAD4 to activate the transcription of target genes, including hepcidin (HAMP).15-17 This pathway is activated in response to iron via BMP618,19 and by BMP2 independently of BMP6.20 Hepcidin suppression during erythropoiesis is mediated at least in part by erythroferrone (ERFE), a protein synthesized in erythroblasts in response to erythropoietin (EPO), downstream of JAK2-STAT5 signaling.21 ERFE inhibits BMP6 and thus downregulates BMP/SMAD signaling in hepatocyes,22-24 thereby suppressing hepcidin.24 ERFE expression is increased in mouse models of β-thalassemia and human thalassemia patients.25,26 Deletion of the gene Erfe (encoding ERFE) in a mouse model of β-thalassemia rescued hepcidin expression and partially decreased the iron accumulation in mice, suggesting that ERFE is a significant contributor to the pathophysiology of the disease.25 Increased hepcidin activity via injection of mini-hepcidin (a synthetic hepcidin analogue) has beneficial effects in mouse models of thalassemia intermedia27 and thalassemia major.28 We therefore sought to develop a therapeutic option to block ERFE activity, thus de-repressing hepcidin suppression and reversing iron overload in iron loading anemias such as β-thalassemia. Here we characterize ERFE binding to different BMPs and show that the N-terminal domain of ERFE is sufficient for hepcidin suppression. We also developed neutralizing anti-ERFE antibodies that prevent ERFE-mediated hepcidin suppression, in vitro and in vivo. Finally, we show that antibodies binding the N-terminal domain of ERFE reduce iron burden and alleviate anemia in a mouse model of β-thalassemia.
Methods
Animal studies and treatments
Animal experiments were undertaken under an approved UK Home Office Project License P5AC0E88C9 with ethics approval from the University of Oxford Animal Welfare and Ethical Review Body. All experiments were performed in male mice. Wild-type male C57BL/6 mice were purchased from Envigo. Embryos from Erfe+/− mice on a mixed Sv129/C57BL/6 background were obtained from the Mutant Mouse Regional Resource Center (MMRRC) at UC Davis (strain B6;129S5-Erfetm1Lex/Mmucd, ID MMRRC:032289-UCD) and backcrossed using marker-assisted accelerated backcrossing yielding ∼99% C57BL/6 background. Heterozygote pairs were mated to generate homozygous animals from which knockout and wild-type colonies were maintained. ERFE knockout mice were used for immunizations. Wild-type and ERFE knockout animals were housed in individually ventilated cages in the Department of Biomedical Services, University of Oxford, and provided access to normal chow (163 ppm of iron, Special Diets Services 801700) and water ad libitum. Hbb(th3/+) mice were obtained from, and then bred and maintained at, The Jackson Laboratories. Age- and sex-matched Hbb(th3/+) mice were shipped to the Pfizer animal facility for experiments and maintained on an iron-sufficient diet (67 ppm of iron, TestDiet 5755). The dietary iron content was lowered in Hbb(th3/+) mice because the higher iron content in standard diet together with ongoing stress erythropoiesis in these mice could lead to unwanted pathology. An iron-sufficient diet was also used in wild-type mice when used as control groups for experiments involving Hbb(th3/+) mice.
For ERFE treatments in wild-type mice, 8-week-old mice were injected intravenously with 100 µg of human ERFE (F2 construct) or saline (vehicle) and euthanized 3 hours after treatment. For antibody testing in EPO-treated mice, wild-type mice were injected intraperitoneally with 200 IU of recombinant human EPO (Bio-Rad) in water, alone or in combination with intravenous injection of anti-ERFE antibodies (5 mg/kg). Two different regimens were used: (1) one simultaneous injection of EPO and antibody and cull 18 hours after treatments; or (2) daily EPO injections for 3 consecutive days, with simultaneous antibody treatment on days 1 and 3, and cull 24 hours after the last EPO injection. In both cases, the control groups used were non-EPO–treated mice (vehicle + IgG antibody control) and EPO-treated with IgG antibody control mice. For antibody testing in Hbb(th3/+) mice, 4-week-old mice were treated intravenously with 5 mg/kg of IgG2A control antibody, anti-ERFE 15.1, or anti-ERFE 20.1 twice a week for 4 or 8 weeks. IgG2A-treated wild-type mice were used as control for baseline levels in the 4 weeks of experiment. After 4 or 8 weeks of treatment, mice were euthanized for analysis of serum and liver iron. Mice were euthanized in increasing carbon dioxide (CO2) concentrations.
Protein production
Expi293F cells (A14527; Thermo Fisher Scientific) were propagated in Expi293 expression medium at 36°C and transfected with intact expression vector DNA (human or mouse ERFE) according to manufacturer’s recommended protocols. After transfection, the cell culture was returned to an 8% CO2 shaking incubator at 36°C. No supplementary tryptone feed was given to these cultures. Conditioned media (CM) were collected 120 hours’ post-transfection. CM were transferred to sterile 1 L Nalgene bottles and centrifuged for 6 minutes at 1800 rpm, 4°C in a Sorvall H-6000A rotor (940g). Clarified CM were collected and filtered by using a Sartopore 2XL 0.8/0.2 μm filtration. ERFE proteins were linked to monoFc29 by using a G4S linker at the N terminus. The recombinant monoFc-huERFE (human) and monoFc-muERFE (murine) were purified in the following manner. All chromatography steps were performed at 4°C. The proteins were captured from CM using MabSelect SuRe (GE Healthcare), washed extensively with calcium-magnesium-free (CMF) phosphate-buffered saline (PBS) pH 7.2, and eluted with a decreasing pH gradient. Fractions containing ERFE were exchanged into low sodium chloride pH 8.0 buffer, loaded onto a Q Sepharose HP column (GE Healthcare), and an increasing sodium chloride gradient elution was performed. A HiLoad Superdex 200 column (GE Healthcare) was used as a final purification step with a mobile phase containing arginine pH 7.0. Fractions with high purity of ERFE were pooled and buffer exchanged into PBS-CMF pH 7.2. Analysis of the ERFE sequence indicates the presence of 2 potential furin cleavage sites, at positions 42 and 212. Constructs were designed based on all the potential furin cleavage variants that could exist assuming cleavage by furin at both sites. This approach resulted in 5 furin cleavage constructs being produced (Figure 2). These constructs were linked to monoFc29 by using a G4S linker at the N terminus. Furin subunit constructs were expressed and purified as previously described. Sequences are available upon request.
Antibody discovery: naive human phage library approach
An antibody scFv phage display library (WyHN5) was used to screen for binders to ERFE. Four rounds of selection with decreasing antigen concentration were conducted by using biotinylated ERFE. Selections were conducted as previously described.30 Antibodies were screened by using an enzyme-linked immunosorbent assay (ELISA) for binding to human, cyno, and murine ERFE. Cross-reactive binders were then assessed in a C2C12 cell line containing a BMP response element reporter fusion to determine their ability to interfere with BMP6 signaling. Antibodies capable of interfering with BMP6 signaling were selected for further characterization and used in homogeneous time-resolved fluorescence (HTRF) assays (Figure 1C). A mouse hybridoma approach was also taken to generate anti-ERFE antibodies as described previously24 ; details are given in the supplemental Methods (available on the Blood Web site).
Antibody production
Transient HEK293 cells expressing anti-ERFE were cultured in FreeStyle 293 medium or Expi293 medium (Thermo Fisher Scientific). These cells were preseeded in a wave bioreactor at a cell density of 1.25 × 106 cells/mL and transfected with polyethylenimine (Polysciences). The wave bioreactors were incubated at 37°C with a rocking rate of 20 rpm for 120 hours before harvest. The CM were centrifuged by using a Sorvall BIOS 16 Bioprocessing Centrifuge (Thermo Fisher Scientific) and filtered with a 0.22 μm filter device before purification. The clarified CM were loaded onto a 5 mL MAbSelect SuRe column (GE Life Sciences) equilibrated with PBS, pH 7.2. The column was washed with 10 column volumes of PBS, pH 7.2 before the protein was step eluted using a low pH buffer. The protein was immediately loaded onto a 320 mL Superdex 200 size exclusion column (GE Life Sciences) equilibrated in PBS, pH 7.2. Peak fractions were pooled and filtered through a 0.2 μm polyethersulfone filter.
Luciferase assay
C2C12 mouse myoblast cells were transfected with a pTal-Luc reporter plasmid in which a synthetic BMP-response element was inserted into the NheI site as previously described.31 Cells were cultured in low bicarbonate Dulbecco’s modified Eagle medium supplemented with 4 mM of L-glutamine, 4.5 g/L of glucose, 100 μg/mL of penicillin-streptomycin, and 10% fetal bovine serum. Cells were plated in a 96-well plate (10 000 cells per well) and treated 24 hours later in 1% fetal bovine serum media containing BMP (2 nM) alone or in combination with a gradient of mouse ERFE concentrations (fourfold dilutions from 0.5 µM) with 2 replicates per condition. Luminescence was measured 24 hours after treatment using the britelite Plus Reporter Gene Assay System (PerkinElmer) and an EnVision Plate Reader (PerkinElmer) following the manufacturer’s instructions.
Surface plasmon resonance
The binding affinities of recombinant human (rh) BMP2 (R&D 355-BM/CF), rhBMP4 (R&D 314-BP/CF), and rhBMP6 (R&D 507-BP/CF) to human ERFE were determined by using a Biacore T200 instrument (GE Healthcare) at 15°C with a collection rate of 10 Hz. The running and sample buffer was 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid pH 7.4, 0.3 M NaCl, 3 mM EDTA, 0.05% P-20. Human ERFE was immobilized onto different flow cells of a CM4 Sensor Chip (GE Healthcare, BR100534) using the Amine Coupling Kit (GE Healthcare, BR-1006-33) according to the manufacturer’s protocol. The immobilization level of human ERFE was 256 resonance units. A twofold dilution series of rhBMP2, rhBMP4, and rhBMP6 and growth differentiation factor-15 (GDF15) with concentrations ranging from 50 nM to 3.13 nM were injected over the sensor surface for 30 seconds at 100 µL/min. The dissociation was monitored for 300 seconds, and the surface was regenerated with 10 mM glycine, pH 1.5. Apparent binding affinities and rate constants were determined by fitting the resulting sensorgram data to a 1:1 Langmuir model in Biacore T200 Evaluation software version 2.0 (GE Healthcare).
Cell culture and treatments
Huh7 cells were cultured in Dulbecco’s modified Eagle medium–high glucose (MilliporeSigma), supplemented with 10% fetal bovine serum (MilliporeSigma), 1% penicillin-streptomycin (MilliporeSigma), and 1% L-glutamine (MilliporeSigma) and maintained at 37°C, 5% CO2. Cells were plated 24 hours before treatments in 24-well cell culture plates. At the time of treatment, cells were washed with PBS, and fresh media were added. Cells were treated with human ERFE (F1-F5 subunits, 1 µg/mL) or full-length mouse ERFE (200 ng/mL) in combination with anti-ERFE antibodies (10 µg/mL), and analyzed 24 hours after treatments. In both cases, non–ERFE-treated cells (saline) were used as control. Each treatment was performed using 3 replicates.
Homogeneous time-resolved fluorescence
A competition HTRF assay32,33 was established to assess whether BMPs could compete with neutralizing anti-ERFE antibodies for binding to the same/overlapping epitope on recombinant ERFE protein, and was performed as previously described.24 Additional details are provided in the supplemental Methods.
Methods for ELISA, RNA isolation, complementary DNA synthesis and quantitative reverse transcription polymerase chain reaction, tissue non-heme iron measurement, blood parameters, serum iron, and transferrin saturation analysis are provided in the supplemental Methods.
Statistical analysis
Statistical analyses were performed by using Prism 7 (GraphPad Software). Statistical significance was assessed by using the Student t test, Mann-Whitney U test, or one-way analysis of variance followed by the Tukey test for multiple comparisons.
Results
ERFE binds to BMP2, BMP4, and BMP6 with different affinities
Previous research from our group described how ERFE inhibits BMP6, thus decreasing hepcidin expression.24 To further characterize this interaction, we performed surface plasmon resonance using the Biacore platform, and compared ERFE binding to BMP2, BMP4, BMP6, and GDF15 (Figure 1A-B). Analysis revealed a high-affinity protein–protein interaction between ERFE and BMP6 (apparent Kd 1.17 nM), consistent with our hypothesis that ERFE acts as a ligand trap for BMP6. Interestingly, we also observed that ERFE binds BMP2 and BMP4, although with lower affinity than BMP6 (16.15 nM and 41.10 nM, respectively). This observation was supported by HTRF data indicating a reduced ability of BMP2 and BMP4 (compared with BMP6) to displace a bound neutralizing antibody from human ERFE (Figure 1C). No binding was detected between ERFE and GDF15, a transforming growth factor β family protein previously proposed to suppress hepcidin. These data show that ERFE directly binds BMP6, the main ligand involved in hepcidin regulation in response to iron, but can also bind BMP2 and BMP4.
![ERFE binds to BMP2, BMP4, and BMP6 with different affinities. (A) Binding kinetics of ERFE to BMP2, BMP4, BMP6, and GDF15 assessed by surface plasmon resonance (Biacore). No binding was observed for GDF15. Full-length human ERFE was immobilized in a CM4 chip. BMPs/GDF15 were tested at different concentrations (3.1-50 nM) for binding to immobilized ERFE and assayed for up to 60 seconds (2 replicates per condition). RU, resonance units. Apparent Kd (dissociation constant) was calculated for binding of BMP2, BMP4, and BMP6 to ERFE (assuming 1:1 binding interaction). (B) Apparent Kd of ERFE to BMP2, BMP4, and BMP6. (C) HTRF assay for detection of binding competition between BMPs (0.1-400 nM) and an anti-ERFE antibody. Unlabeled anti-ERFE antibody (anti-ERFE) and a control IgG antibody were used as positive and negative controls, respectively. Values calculated as %ΔF = [(F665 Sample/F615 Sample) − (F665 Control/F615 Control)/(F665 Control/F615 Control)] × 100. Data plotted as “% Control” represent the background fluorescence energy transfer in wells containing each labeled antibody, in assay buffer, alone.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/8/10.1182_blood.2019003140/5/m_bloodbld2019003140f1.png?Expires=1765884789&Signature=0vUmwzurmaa-E6iE2jP-SdNrP3wNog4VK4zrNAYFkS8zCCSl2R0ZrJytlAigEvyM4UjpkUxu7LFWDu87DFpxxjLmjhBPuS6NsICwmD7w2FCejZQzfXepvDhPKK2dvD9zuIFtNbW3RRcqrYgBkzj~jhScCg1eGaFjUKmwq1oz5uq8VMvi1NjRR8jgMDOn~GgsiOJ6CcVT~Xfb810-LFEP8xWcaRfHnkjxijWirvvB1ErR~ET~uTxxD5tjhiSLpNms42ZbynvQhs8cmW1lOjBbWvq7qOUdvZHUDO9IHIMf-kg6wtdABIY~NjQYrkdoNsRx1HZvbOrpSpDkMnE-y3HyBg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ERFE binds to BMP2, BMP4, and BMP6 with different affinities. (A) Binding kinetics of ERFE to BMP2, BMP4, BMP6, and GDF15 assessed by surface plasmon resonance (Biacore). No binding was observed for GDF15. Full-length human ERFE was immobilized in a CM4 chip. BMPs/GDF15 were tested at different concentrations (3.1-50 nM) for binding to immobilized ERFE and assayed for up to 60 seconds (2 replicates per condition). RU, resonance units. Apparent Kd (dissociation constant) was calculated for binding of BMP2, BMP4, and BMP6 to ERFE (assuming 1:1 binding interaction). (B) Apparent Kd of ERFE to BMP2, BMP4, and BMP6. (C) HTRF assay for detection of binding competition between BMPs (0.1-400 nM) and an anti-ERFE antibody. Unlabeled anti-ERFE antibody (anti-ERFE) and a control IgG antibody were used as positive and negative controls, respectively. Values calculated as %ΔF = [(F665 Sample/F615 Sample) − (F665 Control/F615 Control)/(F665 Control/F615 Control)] × 100. Data plotted as “% Control” represent the background fluorescence energy transfer in wells containing each labeled antibody, in assay buffer, alone.
ERFE binds to BMP2, BMP4, and BMP6 with different affinities. (A) Binding kinetics of ERFE to BMP2, BMP4, BMP6, and GDF15 assessed by surface plasmon resonance (Biacore). No binding was observed for GDF15. Full-length human ERFE was immobilized in a CM4 chip. BMPs/GDF15 were tested at different concentrations (3.1-50 nM) for binding to immobilized ERFE and assayed for up to 60 seconds (2 replicates per condition). RU, resonance units. Apparent Kd (dissociation constant) was calculated for binding of BMP2, BMP4, and BMP6 to ERFE (assuming 1:1 binding interaction). (B) Apparent Kd of ERFE to BMP2, BMP4, and BMP6. (C) HTRF assay for detection of binding competition between BMPs (0.1-400 nM) and an anti-ERFE antibody. Unlabeled anti-ERFE antibody (anti-ERFE) and a control IgG antibody were used as positive and negative controls, respectively. Values calculated as %ΔF = [(F665 Sample/F615 Sample) − (F665 Control/F615 Control)/(F665 Control/F615 Control)] × 100. Data plotted as “% Control” represent the background fluorescence energy transfer in wells containing each labeled antibody, in assay buffer, alone.
The N-terminal domain of ERFE is sufficient to suppress hepcidin
The structure of ERFE shares characteristics with other members of the C1q/tumor necrosis factor–related protein (CTRP) family: a signal peptide, an N-terminal domain containing a collagen-like domain (G-X-Y), and a globular domain homologous to complement protein C1q (Figure 2A). The N-terminal domain is less conserved than the C1q-like domain both between mouse and human orthologs (71% identity overall, 84% within C1q-like region) and also within the human CTRP family as a whole.11 Computational analysis of the human ERFE amino acid sequence using ProP1.0 software predicted 2 furin cleavage sites: RARR at position 42 and RLRR at position 212. To understand which ERFE domain(s) are necessary for ERFE activity, we designed and expressed ERFE subunits based on the predicted furin cleavage sites, and tested these in human hepatoma cells (Huh7). Subunits containing most of the N-terminal domain (F2 and F4) caused hepcidin suppression to a similar extent as subunits containing that region plus the globular C1q (F3) and the full-length protein, indicating that the N-terminal domain is sufficient for ERFE activity (Figure 2B). In the absence of the N terminus, subunits containing the signal peptide (F1) or most of the globular C1q (F5) domain did not affect hepcidin expression. Consistent with this finding, a peptide containing the whole C1q domain also failed to suppress hepcidin (supplemental Figure 1), indicating that this portion of the protein is not required for hepcidin suppressive activity in vitro.
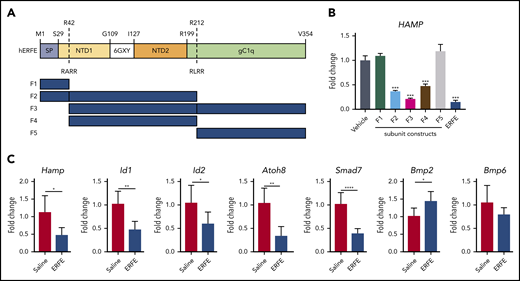
The N-terminal domain of ERFE is sufficient to suppress hepcidin. (A) Structure of human ERFE, containing a signal peptide (SP), N-terminal region (NTD) with a collagen-like domain (6 GXY), and a globular C1q domain (gC1Q). The location of the 2 predicted furin cleavage sites are indicated (RARR and RLRR). ERFE subunits designed and expressed based on predicted furin cleavage sites (F1-F5). (B) Huh7 cells were treated for 24 hours with 1 µg/mL of ERFE subunits F1 to F5, full-length or vehicle, and analyzed for HAMP gene expression. (C) Wild-type mice were treated intraperitoneally with 100 µg of F2 ERFE or saline and analyzed 3 hours after treatment for liver gene expression of 5 BMP target genes (Hamp1, Id1, Id2, Atoh8, Smad7) and Bmp2 and Bmp6. Columns represent mean ± standard deviation. n = 3 replicates per group, panel B; n = 6 mice per group, panel C. *P < .05; **P < .01; ***P < .001; ****P < .0001 using the Student t test.
The N-terminal domain of ERFE is sufficient to suppress hepcidin. (A) Structure of human ERFE, containing a signal peptide (SP), N-terminal region (NTD) with a collagen-like domain (6 GXY), and a globular C1q domain (gC1Q). The location of the 2 predicted furin cleavage sites are indicated (RARR and RLRR). ERFE subunits designed and expressed based on predicted furin cleavage sites (F1-F5). (B) Huh7 cells were treated for 24 hours with 1 µg/mL of ERFE subunits F1 to F5, full-length or vehicle, and analyzed for HAMP gene expression. (C) Wild-type mice were treated intraperitoneally with 100 µg of F2 ERFE or saline and analyzed 3 hours after treatment for liver gene expression of 5 BMP target genes (Hamp1, Id1, Id2, Atoh8, Smad7) and Bmp2 and Bmp6. Columns represent mean ± standard deviation. n = 3 replicates per group, panel B; n = 6 mice per group, panel C. *P < .05; **P < .01; ***P < .001; ****P < .0001 using the Student t test.
Previous data from our group have shown that full-length ERFE directly suppresses hepcidin and BMP target genes in mice 3 hours after an intravenous injection with recombinant protein. To corroborate our in vitro data on functionality of ERFE subunits, we treated mice with F2 ERFE, containing the signal peptide and the N-terminal domain but not the globular C1q domain. Analysis at 3 hours after treatment showed suppression of Hamp and other BMP-target genes (Id1, Id2, Atoh8, and Smad7), with no suppression of Bmp2 or Bmp6 (Figure 2C). These data confirm our previous findings that ERFE suppresses hepcidin and BMP signaling in vivo and show that the N-terminal domain is sufficient for ERFE activity.
Neutralizing anti-ERFE antibodies bind to the N-terminal region of ERFE
Abnormally high circulating ERFE levels are associated with increased severity of β-thalassemia,26 causing persistent hepcidin suppression that leads to iron accumulation. To neutralize excess ERFE, we developed anti-ERFE antibodies by immunizing ERFE knockout mice with full-length protein. After antibody purification, we tested the binding capacity of the antibodies to different regions of ERFE by using an ELISA and identified antibodies that exclusively bind to the N-terminal domain (monoclonal antibodies [mAbs] 15.1 and 20.1) or globular C1q domain (mAb 28.1) (Figure 3A). HTRF analysis showed that binding of an anti–N terminus antibody to full-length ERFE is not strongly inhibited by anti–C terminus antibody and that binding of anti–N terminus antibody to ERFE, but not binding of anti–C terminus antibody, is potently and dose dependently inhibited by BMP6 (Figure 3B). These findings show that the BMP6 binding site is fully or partially contained within the N terminus. This is consistent with a proposed mechanism for ERFE action in which the N-terminal domain of ERFE binds BMP6 and acts as a ligand trap to suppress BMP/SMAD signaling and hepcidin expression.
![Neutralizing anti-ERFE antibodies bind to the N-terminal region of ERFE. (A) Anti-ERFE antibodies (15.1, 20.1, and 28.1) were assayed at different concentrations (10−5-102 nM) in ELISA plates coated with full-length ERFE, F4 (N-terminal domain region), and the globular C1q domain. (B) HTRF assay for detection of binding competition using cryptate-labeled anti-ERFE antibodies (20.1 and 28.1) and BMP6 (0.1-200 nM) and unlabeled antibody as positive control. Values calculated as %ΔF = [(F665 Sample/F615 Sample) − (F665 Control/F615 Control)/(F665 Control/F615 Control)] × 100. Data plotted as “% Control” represent the background fluorescence energy transfer in wells containing each labeled antibody, in assay buffer, alone. (C) Huh7 cells were treated for 24 hours with 200 ng/mL of murine ERFE alone or in combination with 10 µg/mL of anti-ERFE antibodies 15.1 and 20.1, and analyzed for HAMP gene expression. Bars represent mean ± standard deviation. n = 3 replicates per group. ***P < .001; ****P < .0001 using the Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/8/10.1182_blood.2019003140/5/m_bloodbld2019003140f3.png?Expires=1765884789&Signature=CoOHr0jcgOAPIvAj3scUOcbmaNyp5PqB99BBo9XD-eyhfpVJ0Za5JH0JmFTpbvdoj3YaWimEbpxdWqjyWCjZEVmISDya798JiE8AhIw3XQnIvubMtCIoyZEIP05o69bO4NXBRsfUxUWWyvrRL7RY6r~DueL0JNXai7DPT3pbOL5iTbVHML1~yrAExObXV6nIOxcwhSnjZ5PhCMFefb242YwEbio7Hxfx7Rejj~NmeqY2iuS1vC1pRwIgxhgbIBFBi6UFfGKtfzwzwFVeWJ0HFyo0TRArkP1iRYH7dtOSkte38v1vMlRuDqrDDjLKQiu0KNr8w2lDkugNjTr1kzd-0A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Neutralizing anti-ERFE antibodies bind to the N-terminal region of ERFE. (A) Anti-ERFE antibodies (15.1, 20.1, and 28.1) were assayed at different concentrations (10−5-102 nM) in ELISA plates coated with full-length ERFE, F4 (N-terminal domain region), and the globular C1q domain. (B) HTRF assay for detection of binding competition using cryptate-labeled anti-ERFE antibodies (20.1 and 28.1) and BMP6 (0.1-200 nM) and unlabeled antibody as positive control. Values calculated as %ΔF = [(F665 Sample/F615 Sample) − (F665 Control/F615 Control)/(F665 Control/F615 Control)] × 100. Data plotted as “% Control” represent the background fluorescence energy transfer in wells containing each labeled antibody, in assay buffer, alone. (C) Huh7 cells were treated for 24 hours with 200 ng/mL of murine ERFE alone or in combination with 10 µg/mL of anti-ERFE antibodies 15.1 and 20.1, and analyzed for HAMP gene expression. Bars represent mean ± standard deviation. n = 3 replicates per group. ***P < .001; ****P < .0001 using the Student t test.
Neutralizing anti-ERFE antibodies bind to the N-terminal region of ERFE. (A) Anti-ERFE antibodies (15.1, 20.1, and 28.1) were assayed at different concentrations (10−5-102 nM) in ELISA plates coated with full-length ERFE, F4 (N-terminal domain region), and the globular C1q domain. (B) HTRF assay for detection of binding competition using cryptate-labeled anti-ERFE antibodies (20.1 and 28.1) and BMP6 (0.1-200 nM) and unlabeled antibody as positive control. Values calculated as %ΔF = [(F665 Sample/F615 Sample) − (F665 Control/F615 Control)/(F665 Control/F615 Control)] × 100. Data plotted as “% Control” represent the background fluorescence energy transfer in wells containing each labeled antibody, in assay buffer, alone. (C) Huh7 cells were treated for 24 hours with 200 ng/mL of murine ERFE alone or in combination with 10 µg/mL of anti-ERFE antibodies 15.1 and 20.1, and analyzed for HAMP gene expression. Bars represent mean ± standard deviation. n = 3 replicates per group. ***P < .001; ****P < .0001 using the Student t test.
We next tested the capacity of the antibodies to neutralize ERFE in vitro. As expected, Huh7 cells treated with full-length recombinant mouse ERFE suppressed hepcidin (Figure 3C). When the same conditions were used in the presence of an anti-ERFE mAb (15.1 or 20.1), hepcidin suppression was absent (15.1) or severely blunted (20.1), indicating that both antibodies targeting the N-terminal domain neutralize ERFE.
Antibodies binding ERFE N-terminal domain block hepcidin suppression in EPO-treated mice
ERFE expression and production in erythroblasts is stimulated by EPO; under basal physiological conditions, the messenger RNA is lowly expressed by those cells (and there are fewer such cells). To test our antibodies in a model of increased erythropoiesis (leading to increased ERFE), we injected wild-type mice with 200 IU of EPO plus an antibody control (IgG2A) or 1 of the 2 neutralizing antibodies described earlier, and analyzed the mice 18 hours after treatment. In the presence of control IgG2A, EPO suppressed hepcidin (Figure 4A), leading to lower circulating hepcidin protein (Figure 4B). Simultaneous injection of EPO and anti-ERFE antibody prevented hepcidin suppression and rescued circulating hepcidin to the levels observed in non–EPO-treated mice. To test the functional impact of ERFE neutralization on erythropoietic output, we increased the dose to 3 EPO injections on 3 consecutive days, with simultaneous injection of anti-ERFE antibody on days 1 and 3, and analysis of blood parameters on day 4. In these conditions, a moderate (15.1) or a significant (20.1) elevation in Hamp expression was observed compared with EPO-treated mice (supplemental Figure 2A), with decreased serum iron and unchanged liver iron (supplemental Figure 2B). Various blood parameters were increased by EPO treatment (hemoglobin, hematocrit, mean corpuscular volume, and red blood cell distribution width). These increases were partially prevented by treatment with anti-ERFE (supplemental Figure 2C). Thus, inhibition of ERFE affects hepcidin and erythropoiesis output in response to EPO.

Antibodies binding ERFE N-terminal domain block hepcidin suppression in EPO-treated mice. Eight-week-old wild-type male mice were treated intraperitoneally with 200 IU of EPO in combination with intravenous injection of an IgG2A antibody control, anti-ERFE 15.1, or anti-ERFE 20.1 (or vehicle alone instead of EPO for analysis of baseline values). Mice were killed and analyzed 18 hours after treatment for assessment of Hamp expression (A) and serum hepcidin (B). n = 3, vehicle; n = 5-6, EPO-treated mice. *P < .05; **P < .01; ***P < .001; ****P < .0001 using the Student t test.
Antibodies binding ERFE N-terminal domain block hepcidin suppression in EPO-treated mice. Eight-week-old wild-type male mice were treated intraperitoneally with 200 IU of EPO in combination with intravenous injection of an IgG2A antibody control, anti-ERFE 15.1, or anti-ERFE 20.1 (or vehicle alone instead of EPO for analysis of baseline values). Mice were killed and analyzed 18 hours after treatment for assessment of Hamp expression (A) and serum hepcidin (B). n = 3, vehicle; n = 5-6, EPO-treated mice. *P < .05; **P < .01; ***P < .001; ****P < .0001 using the Student t test.
Anti-ERFE antibodies decrease iron levels and alter blood parameters in thalassemic mice
To assess the capacity of anti-ERFE antibodies as a potential therapeutic option for preventing iron overload in β-thalassemia, we initially tested both anti–N terminus Erfe antibodies in Hbb(th3/+) mice, a model of thalassemia intermedia. Mice were treated with anti-ERFE or IgG2A control biweekly for 4 weeks, starting at 4 weeks of age. During this period, these mice accumulated iron associated with suppressed hepcidin and increased ERFE expression,25 characteristics observed in human patients. Compared with wild-type mice, IgG2A-treated thalassemic mice had lower serum iron and transferrin saturation (Figure 5B), increased iron accumulation in the liver and spleen (Figure 5C), and a higher spleen/body weight ratio (Figure 5D). Treatment with both anti-ERFE antibodies caused a further decrease in serum iron, lowered the spleen/body weight ratio, and antibody 15.1 partially prevented iron accumulation in the liver. However, serum hepcidin levels were not significantly different between groups (Figure 5A). Thus, the lower levels of serum and liver iron in anti–ERFE-treated mice were not paralleled with decreased hepcidin, indicating that hepcidin might be relatively increased by anti-ERFE treatment. Concurrently, treatment with anti-ERFE antibodies increased the number of red blood cells, hemoglobin concentration, and hematocrit, while decreasing the number of reticulocytes and the red blood cell distribution width (Figure 5E).
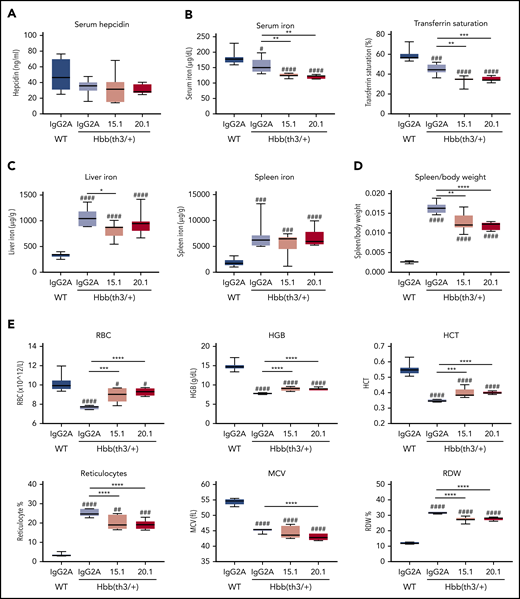
Antibodies targeting the N-terminal domain of ERFE decrease iron levels and alter blood parameters in thalassemic mice. Four-week-old male Hbb(th3/+) mice were treated intravenously with 5 mg/kg of IgG2A control antibody, anti-ERFE 15.1, or anti-ERFE 20.1, twice a week for 4 weeks. IgG2A-treated wild-type (WT) mice were used as control for basal levels. After 4 weeks of treatment, mice were killed for analysis of serum hepcidin (A), serum iron and transferrin saturation (B), liver and spleen non-heme iron (C), spleen to body weight ratio (D), and blood parameters (E). *P < .05; **P < .01; ***P < .001; ****P < .0001 using one-way analysis of variance followed by the Tukey test for differences between IgG2A-treated Hbb(th3/+) mice and anti–ERFE-treated mice. #P < .05, ##P < .01, ###P < .001, ####P < .0001 using one-way analysis of variance followed by the Tukey test for differences between WT mice and Hbb(th3/+) mice. HCT, hematocrit; HGB, hemoglobin; MCV, mean corpuscular volume; RBC, red blood cells; RDW, red blood cell distribution width. n = 5-8 mice per group.
Antibodies targeting the N-terminal domain of ERFE decrease iron levels and alter blood parameters in thalassemic mice. Four-week-old male Hbb(th3/+) mice were treated intravenously with 5 mg/kg of IgG2A control antibody, anti-ERFE 15.1, or anti-ERFE 20.1, twice a week for 4 weeks. IgG2A-treated wild-type (WT) mice were used as control for basal levels. After 4 weeks of treatment, mice were killed for analysis of serum hepcidin (A), serum iron and transferrin saturation (B), liver and spleen non-heme iron (C), spleen to body weight ratio (D), and blood parameters (E). *P < .05; **P < .01; ***P < .001; ****P < .0001 using one-way analysis of variance followed by the Tukey test for differences between IgG2A-treated Hbb(th3/+) mice and anti–ERFE-treated mice. #P < .05, ##P < .01, ###P < .001, ####P < .0001 using one-way analysis of variance followed by the Tukey test for differences between WT mice and Hbb(th3/+) mice. HCT, hematocrit; HGB, hemoglobin; MCV, mean corpuscular volume; RBC, red blood cells; RDW, red blood cell distribution width. n = 5-8 mice per group.
To further assess the effects of an anti-ERFE antibody, we increased the dosing period to 8 weeks, which led to more pronounced effects: anti-ERFE 15.1 treatment caused an increase in hepatic hepcidin messenger RNA expression, a decrease in liver iron, and thus an increased Hamp to liver iron content ratio (Figure 6A-C). Serum iron was decreased, and spleen iron concentration was not significantly different but a reduction in spleen/body ratio was observed (Figure 6D-F). This scenario indicates an amelioration of splenomegaly, a major feature of β-thalassemia that is recapitulated in Hbb(th3/+) mice. Increased red blood cells, hemoglobin, and hematocrit, and fewer reticulocytes, were also evident (Figure 6G). Overall, anti-ERFE antibodies decreased iron accumulation in thalassemic mice and improved anemia.
![Antibodies targeting the N-terminal domain of ERFE increase hepcidin expression and ameliorate anemia in thalassemic mice. Four-week-old male Hbb(th3/+) mice were treated intravenously with 5 mg/kg of IgG2A control antibody, or anti-ERFE 15.1, twice a week for 8 weeks. After 8 weeks of treatment, mice were killed for analysis of liver hepcidin messenger RNA expression (A), liver non-heme iron (B), Hamp to liver non-heme iron content (liver iron content [LIC]) ratio (C), serum iron (D), spleen non-heme iron (E), spleen to body weight (F), and blood parameters (G). *P < .05; **P < .01; ***P < .001 using the Mann-Whitney U test. HCT, hematocrit; HGB, hemoglobin; RBC, red blood cells. n = 6, IgG2A group; n = 9, the 15.1 group.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/8/10.1182_blood.2019003140/5/m_bloodbld2019003140f6.png?Expires=1765884789&Signature=l-YxoecFSC1NWts0kC-Vz~eT1-nUH1z4W776BM6dxkLRLPbeL7rl0lzj9SzmmrSF3bPOdowPcw4xYlbSPN3P1A1mBtCqcM8-3JBegKdV4m6bgxsfmadTmiK-K2AZXdnwlYDxiJk9n-vnEW~y8Mfuogj~MO~aD7zAbeNIixrYeW31gtGsXGh8fFaQMMozTnkth2RCe1szlXQRdATkib6NXb2XHC-S3oJhm-4H79aqUsGp4YtPLHKCX~WZIj-GQduZnvDkybb9OxsHRzJxnZLwtQmQ7b70CBkVl88UeNn2puP7FMdpkSLearzvnWR5vHP4AS0kRHsAm6jj-SS-4019Ug__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Antibodies targeting the N-terminal domain of ERFE increase hepcidin expression and ameliorate anemia in thalassemic mice. Four-week-old male Hbb(th3/+) mice were treated intravenously with 5 mg/kg of IgG2A control antibody, or anti-ERFE 15.1, twice a week for 8 weeks. After 8 weeks of treatment, mice were killed for analysis of liver hepcidin messenger RNA expression (A), liver non-heme iron (B), Hamp to liver non-heme iron content (liver iron content [LIC]) ratio (C), serum iron (D), spleen non-heme iron (E), spleen to body weight (F), and blood parameters (G). *P < .05; **P < .01; ***P < .001 using the Mann-Whitney U test. HCT, hematocrit; HGB, hemoglobin; RBC, red blood cells. n = 6, IgG2A group; n = 9, the 15.1 group.
Antibodies targeting the N-terminal domain of ERFE increase hepcidin expression and ameliorate anemia in thalassemic mice. Four-week-old male Hbb(th3/+) mice were treated intravenously with 5 mg/kg of IgG2A control antibody, or anti-ERFE 15.1, twice a week for 8 weeks. After 8 weeks of treatment, mice were killed for analysis of liver hepcidin messenger RNA expression (A), liver non-heme iron (B), Hamp to liver non-heme iron content (liver iron content [LIC]) ratio (C), serum iron (D), spleen non-heme iron (E), spleen to body weight (F), and blood parameters (G). *P < .05; **P < .01; ***P < .001 using the Mann-Whitney U test. HCT, hematocrit; HGB, hemoglobin; RBC, red blood cells. n = 6, IgG2A group; n = 9, the 15.1 group.
Discussion
The existence of an erythroid regulator that increases iron availability in response to erythropoietic demand has long been hypothesized,34 and the discovery of ERFE provided a strong candidate to fill that role.21 Increasing hepcidin activity is a therapeutic goal for iron loading anemias such as β-thalassemia, and inhibiting ERFE activity is one potential method of achieving this (among others27,35-38 ); antibodies that specifically bind ERFE were recently described (https://patents.google.com/patent/WO2018027184A1/en). We generated ERFE-neutralizing antibodies that rescue ERFE-mediated hepcidin suppression both in vitro (Figure 3C) and in mice injected with EPO (Figure 4, supplemental Figure 2), and which decreased serum and liver iron in thalassemic mice (Figures 5 and 6). Liver iron in anti–ERFE-treated Hbb(th3/+) mice was not reduced to the levels observed in wild-type mice, which may be due to iron accumulation before administration of anti-ERFE and/or because the increase in hepcidin mediated by anti-ERFE is not sufficient to completely inhibit dietary absorption. Even though the Hbb(th3/+) mice recapitulate many of the features of the human disease (low hemoglobin, increase in reticulocyte numbers, aberrant erythrocyte morphology, and tissue iron overload39,40 ), mice normalize hepcidin expression after 12 weeks of age, in contrast to human patients who present chronic hepcidin suppression throughout adulthood.41-43 For this reason, we hypothesized that neutralizing ERFE in human β-thalassemia might have a more pronounced impact than in mice. Concurrent with the decrease in iron, we observed an amelioration of ineffective erythropoiesis with an increase in the number of red blood cells, hemoglobin, and hematocrit, with a decreased number of reticulocytes, mean corpuscular volume, and cell distribution width. Based on previous studies by others, we can speculate that iron restriction may induce a reduction in heme synthesis to lower the amount of α-globin, thus equilibrating α and β chains, and potentially reducing reactive oxygen species.44,45
We have previously shown that ERFE inhibits BMP6 and suppresses hepcidin expression.24 The current study, using Biacore, shows that the direct BMP6–ERFE interaction is of high affinity (∼1.17 nM), which contrasts with the estimated lower affinity of a TfR2-BMP interaction (∼400 nM).46 The development of anti-ERFE antibodies was also used as a tool to further investigate the ERFE mechanism of action. Using HTRF as a competition assay between anti-ERFE mAbs and BMP6, we show that BMP6 binds to a region of ERFE contained within the N-terminal domain (Figure 3B). Consistent with this notion, the ERFE N-terminal domain was sufficient to suppress hepcidin expression both in vitro and in mice (Figure 2). We therefore showed that ERFE-mediated hepcidin suppression is caused by the N-terminal domain binding to BMP6. The N-terminal region is generally less conserved than the C1q-like domain (which mediates multimerization) and may confer specific activities upon individual CTRP family members.
In addition to BMP6, we have previously shown that ERFE binds BMP5 and BMP7, which are closely related to BMP6 but are not currently regarded as regulators of iron metabolism in vivo. Although BMP7 does not have a described physiological role in iron homeostasis, the ability of ERFE to bind this BMP may be relevant in situations in which lack of BMP6 is compensated by an increase in BMP7 expression.47 Here, we expanded our analysis to BMP2 and BMP4. BMP2 regulates liver hepcidin expression independently of BMP6,20 and BMP4 is structurally related but its physiological relevance with respect to hepcidin regulation is not yet clear. Interestingly, we observed that ERFE does bind these 2 BMPs, although with significantly lower affinity than binding to BMP6 (Figure 1). ERFE does not strongly impair the ability of BMP2 to stimulate hepcidin synthesis in vitro24 but could conceivably influence BMP2 activity in vivo in the absence of BMP6. We recently showed that ERFE can inhibit BMP2 signaling in subcutaneous abdominal preadipocytes.48 Furthermore, ERFE may modulate bone metabolism in β-thalassemia by a mechanism that may involve BMPs49 ; further research is needed to investigate the role of Erfe beyond hepcidin regulation. The relative inability of ERFE to functionally impair BMP2-induced hepcidin induction compared with BMP6, despite ERFE binding BMP2 and influencing BMP signaling in non-hepatocyte cell types, may be related to structural variation in different BMPs17 and/or differences in how BMP-ERFE complexes interact with available receptor/coreceptor combinations on distinct cell types. Structural analysis of ERFE and BMPs and their receptors is required to further investigate this issue. GDF15, previously proposed to control hepcidin expression in response to erythropoiesis,50 did not bind ERFE, indicating that not all members of the transforming growth factor β superfamily are affected by ERFE.
In conclusion, by identifying the active region of ERFE and the binding affinity to different BMPs, we increased our understanding of ERFE mechanism of action. We describe the development of ERFE N terminus–targeted antibodies that neutralize ERFE-mediated hepcidin suppression and ameliorate the iron-loaded phenotype in a mouse model of β-thalassemia, indicating their potential therapeutic utility to treat this disease.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Elizabeth DiBlasio Smith and Chris Corcoran for contributions to protein purification; Darren Ferguson, Caryl Meade, and Ashley Schwab for contributions to antibody production; Jonathon Merrill, Jordan Tanner, and Roo Bhasin for contributions to the techniques involving mice; and Sarah Gooding for useful advice.
This work was funded by a Pfizer-Sponsored Rare Disease Consortium Award, the Medical Research Council UK (MRC Human Immunology Unit core funding MC_UU_12010/10 to H.D.). S.J.D. is a Jenner Investigator, a Lister Institute Research Prize Fellow, and a Wellcome Trust Senior Fellow (106917/Z/15/Z).
Authorship
Contribution: J.A., N.F., K.M., S.B., A.E.A., S.-R.P., D.D.P., J.E.M., E.L., O.C., M.L., S.J.D., R.J., and H.D. designed research; J.A., N.F., K.M., A.S., M.S.T., D.Q., S.B., P.J.L., J.N.F., A.E.A., and S.-R.P. performed research; J.A., N.F., K.M., S.B., A.S., M.S.T., P.J.L., J.N.F., A.E.A., and S.-R.P. collected data; J.A., N.F., K.M., A.S., S.B., O.C., M.L., S.J.D., R.J., and H.D. analyzed and interpreted data; J.A. and N.F. performed statistical analysis; and J.A., N.F., R.J., and H.D. wrote the manuscript.
Conflict-of-interest disclosure: This work was supported in part by funding from Pfizer to J.A., K.M., D.Q., S.J.D., and H.D. N.F., A.S., S.B., E.L., M.S.T., D.D.P., O.C., M.L., J.E.M., and R.J. are employed by Pfizer. N.F., O.C., R.J., J.A., K.M., S.J.D., and H.D. are named inventors on a patent application currently under evaluation. The remaining authors declare no competing financial interests.
Correspondence to: Hal Drakesmith, MRC Human Immunology Unit, MRC Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, OX3 9DS, United Kingdom; e-mail: alexander.drakesmith@imm.ox.ac.uk.
