

Key Points
Complement activation is associated with antiphospholipid antibody–induced thrombotic events.
Patients with catastrophic APS harbor rare germline mutations in complement regulatory genes.

Abstract
The antiphospholipid syndrome (APS) is characterized by thrombosis and/or pregnancy morbidity in the presence of antiphospholipid antibodies, including anti-β2-glycoprotein-I (anti-β2GPI), that are considered central to APS pathogenesis. Based on animal studies showing a role of complement in APS-related clinical events, we used the modified Ham (mHam) assay (complement-dependent cell killing) and cell-surface deposition of C5b-9 to test the hypothesis that complement activation is associated with thrombotic events in APS. A positive mHam (and corresponding C5b-9 deposition) were present in 85.7% of catastrophic APS (CAPS), 35.6% of APS (and 68.5% of samples collected within 1 year of thrombosis), and only 6.8% of systemic lupus erythematosus (SLE) sera. A positive mHam assay was associated with triple positivity (for lupus anticoagulant, anticardiolipin, and anti-β2GPI antibodies) and recurrent thrombosis. Patient-derived anti-β2GPI antibodies also induced C5b-9 deposition, which was blocked completely by an anti-C5 monoclonal antibody, but not by a factor D inhibitor, indicating that complement activation by anti-β2GPI antibodies occurs primarily through the classical complement pathway. Finally, patients with CAPS have high rates of rare germline variants in complement regulatory genes (60%), compared with patients with APS (21.8%) or SLE (28.6%) or normal controls (23.3%), and have mutations at a rate similar to that of patients with atypical hemolytic uremic syndrome (51.5%). Taken together, our data suggest that anti-β2GPI antibodies activate complement and contribute to thrombosis in APS, whereas patients with CAPS have underlying mutations in complement regulatory genes that serve as a “second hit,” leading to uncontrolled complement activation and a more severe thrombotic phenotype.
Introduction
Antiphospholipid syndrome (APS) is an acquired thrombophilia characterized by venous or arterial thrombosis and/or obstetrical morbidity with persistently positive antiphospholipid (aPL) antibodies, including lupus anticoagulant (LA), anticardiolipin antibody (aCL), and anti-β2-glycoprotein-I (β2GP-I).1 Anti-β2GPI antibodies are considered the primary pathogenic antibody in APS.2-4 The mechanisms by which aPLs induce thrombosis are unclear. Multiple mechanisms have been proposed, including inhibition of the natural anticoagulant and fibrinolytic systems5-8 ; activation of vascular cells, including endothelial cells,9 platelets,10 and monocytes11 ; procoagulant effects of extracellular vesicles12 ; disruption of the annexin-A5 shield on cellular surfaces13 ; and complement activation.14-20 The lack of a unifying mechanism likely reflects heterogeneity in pathogenic antibodies and disease biology.
Distinguishing benign from pathogenic aPLs is a major gap in APS care and research. The presence of an LA21,22 and triple positivity (presence of LA, aCL, and anti-β2GPI antibody)23,24 are strong predictors of thrombotic risk in APS at a population, but not an individual, level. Predicting which patients with aPLs are at risk of a first or subsequent thrombotic event remains challenging.25 Long-term anticoagulation with a vitamin K antagonist remains the standard of care for thrombotic APS.26 A severe form of APS, characterized by widespread thrombosis and multiorgan failure developing over less than a week, termed catastrophic APS (CAPS), affects a subset (∼1%) of APS patients. CAPS often presents as a thrombotic microangiopathy (TMA) and has a fulminant course with >40% mortality, despite the best available therapy,26,27 indicating a need for therapies beyond anticoagulation.26
Complement inhibition has emerged as an attractive therapeutic strategy based on evidence of complement activity in patients with APS, murine models that indicate a critical role of complement in aPL-mediated thrombosis14-17 and obstetric18-20 complications, and reports of the efficacy of terminal complement inhibition with eculizumab in patients with refractory thrombotic APS28 and CAPS.29-32 Increased complement activation products, including C5b-9,33 fragment Bb, and C3a,34,35 have been observed in sera of patients with APS; however, the association of serologic evidence of complement activation with thrombotic events is inconsistent.35,36
In this prospective study, we investigated complement activity via cell surface deposition of C5b-9 and complement-dependent cell killing (the modified Ham [mHam] assay) using patient sera and showed that complement activation is associated with thrombotic events in APS. In addition, we demonstrated complement activation by patient-derived purified anti-β2GPI in vitro. Finally, targeted sequencing was performed to test the hypothesis that CAPS is associated with rare germline variants in complement regulatory genes, serving as a “second hit” that accounts for the more severe phenotype observed in this disease.
Methods
Patients and samples
From January 2015 through June 2019, we prospectively recruited patients with thrombotic APS, CAPS, and systemic lupus erythematosus (SLE) from the Johns Hopkins Complement Associated Disorders Registry and the Hopkins Lupus Cohort.37 Patients with CAPS were also recruited from the hematology services at the Cleveland Clinic (Cleveland, OH) and McMaster University (Hamilton, ON, Canada). Patients were diagnosed with thrombotic APS based on International Society on Thrombosis and Hemostasis criteria, including ≥1 clinical episode of arterial, venous, or small-vessel thrombosis and the presence of LA, aCL antibody of the IgG/IgM isotype, or anti-β2GPI antibody of the IgG/IgM isotype detected on at least 2 occasions at least 12 weeks apart.1 Patients were classified as single, double, or triple positive, based on positive assays for 1, 2, or all 3 of LA and anti-β2GPI and aCL antibodies. Patients with recurrent thrombosis confirmed by imaging at any time before enrollment or during follow-up in the registry were classified as having recurrent thrombosis. For patients with multiple tests for aPLs, we used the tests drawn closest to the study sample. CAPS was diagnosed according to international consensus criteria including involvement of 3 or more organs, development of manifestations within a period of a week, histologic confirmation of small vessel thrombosis, and laboratory confirmation of the presence of aPLs.38 The diagnosis of definite CAPS requires all 4 criteria, whereas probable CAPS is diagnosed if 3 criteria are met (when tissue biopsy is not obtained, laboratory testing cannot be repeated because of death, or multiorgan thrombosis develops over more than a week but less than a month, despite anticoagulation).38 We included patients with both definite and probable CAPS, because biopsies to confirm small-vessel thrombosis are commonly omitted in critically ill patients who otherwise meet criteria for CAPS, and outcomes of patients with probable CAPS are comparable to those of patients with definite CAPS.39,40 SLE was diagnosed according to the Systemic Lupus International Collaborating Clinics Criteria.41
For genetic analysis, we used DNA samples from healthy pregnant women recruited from the obstetrics clinics at Johns Hopkins Hospital as a control cohort, as well as patients with atypical hemolytic uremic syndrome (aHUS) as a reference cohort for validation of our custom sequencing panel. aHUS was diagnosed if the first manifestation of the syndrome met the following criteria: (1) platelet count <100 × 109/L, (2) serum creatinine >2.25 mg/dL, and (3) ADAMTS13 activity >10%. These criteria have been used in previous studies to clinically differentiate patients with aHUS.42
Blood was collected by venipuncture in serum separation tubes and was immediately centrifuged at 4°C. Serum was separated and stored at −80°C. Whole blood was used to generate genomic DNA for targeted gene sequencing with a Qiagen DNeasy Blood & Tissue Kit. All samples and data were de-identified and coded after collection. Samples were obtained from patients enrolled in existing registries or were submitted by clinical centers as part of diagnostic testing. This study was approved by the Institutional Review Board at Johns Hopkins University, and was conducted in accordance with the Declaration of Helsinki.
Patient-derived anti-β2GPI antibodies
Anti-β2GPI antibodies from 2 patients were affinity purified on a column of Affigel HZ to which purified human β2GP-I was coupled (Bio-Rad Laboratories, Hercules, CA), as previously described.43 IgG purity was assessed by reduced sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Complement activation (C5b-9 deposition and complement-dependent cell killing by mHam analysis) induced by patient-derived anti-β2GPI antibodies was tested by adding patient-derived anti-β2GPI antibodies to normal human serum (NHS; Complement Technology Inc).
mHam assay
The mHam assay has been validated to assess complement activation in patient serum and was performed as described previously.42,44 In brief, PIGA− TF-1 cells were cultured at 500 000 cells per mL and passaged daily. The cells were washed with phosphate-buffered saline (PBS) and plated in 96-well plates in gelatin veronal buffer with Ca and Mg (GVB++; B102; Complement Technology Inc) at a density of 6700 cells per well in triplicate. Test serum (20 µL at 1:5 dilution) was added to cells and incubated at 37°C for 45 min with constant shaking. After incubation, the cells were washed with PBS and incubated with WST-1 cell proliferation reagent (Roche, Basel, Switzerland) at a dilution of 1:10 for 2 h at 37°C. Absorbance was measured in a plate reader (ELX808; BioTek, Winooski, VT) at 450 nm with a reference wavelength of 630 nm. The percentage of live cells was calculated as the ratio of absorbance of the sample to its heat-inactivated control multiplied by 100. The percentage of nonviable cells (100 − percentage of viable cells) is a measure of complement activation. Based on prior experiments, 20% nonviable cells (cell killing) is established as the threshold for a positive test.42,44 All assays were performed in triplicate for replication.
As a positive control for the mHam, Shiga toxin 145 (SML0562; Sigma-Aldrich, St. Louis, MO) 10 µg/mL was added to NHS and incubated at 37°C for 15 min, followed by the addition of the cells. Heat-inactivated serum was used as an internal negative control for each sample. To confirm complement dependence of cell killing, we performed the mHam assay after adding an anti-C5 monoclonal antibody (10 µg; Alexion Pharmaceuticals). We also performed the assays with a small-molecule factor D inhibitor46 (0.33 µM; ACH-4471; Achillion Pharmaceuticals) to evaluate the contribution of the alternative vs classic/lectin pathways.
Flow cytometry for C5b-9 deposition
Flow cytometry was performed to evaluate cell surface deposition of C5b-9 on PIGA− TF1 cells. Cells were seeded in V-bottom 96-well plates (1.2 × 105 cells per well) either in GVB0·MgEGTA (pH 6.4) buffer for alternative pathway activation or GVB++ (pH 7.4) buffer for all complement pathway activation, followed by addition of 20 µL APS patient serum (or patient-derived aPL added to NHS). The reaction was incubated for 15 min at 37°C with constant shaking, and stopped by adding fluorescence-activated cell sorting buffer supplemented by 1% bovine serum albumin (BSA) and 15 mM EDTA in PBS. Cell pellets were separated by centrifugation at 1900 rpm for 3 min at room temperature and then stained with anti-C5b-9 monoclonal primary antibody (1:50; sc-58935; Santa Cruz Biotechnology) for 30 min, followed by Alexa 647-conjugated secondary antibody (1:500; Ab172325; Abcam). C5b-9 deposition was measured by a BD FACSCalibur and analyzed using FlowJo software, version 10.5.3. NHS with EDTA was used as a negative control for flow cytometry assays. We evaluated the effect of adding anti-C5 mAb (10 µg) and ACH-4471 (0.33 µM) after C5b-9 deposition.
Targeted sequencing
Genomic DNA was quantitated using the Qiagen Qubit fluorometric assay, and 50 ng of DNA was used as input for targeted sequencing with a custom Ampliseq panel (Illumina). Amplicons were designed to cover all exons of 15 genes with known function related to complement activation and regulation (CFH, CFB, CFI, CFD, CFP, CFHR1, CFHR2, CFHR3, CFHR4, CFHR5, C3, CD46 (MCP), THBD, CR1, and DGKE). Complete methods are provided in the supplemental Data, available on the Blood Web site.
Data analysis
Data were summarized as counts (percentage) and medians (Q25% to 75%) or mean (± standard deviation) for categorical and continuous variables, respectively. We used the χ2 test to compare the proportion of patients with positive mHam in the following groups: (1) SLE, APS, and CAPS; (2) single-, double-, and triple-positive APS; and (3) with and without a history of thrombosis. We used the paired Student t test to compare C5b-9 deposition in patient sera with their own heat-inactivated negative controls. We used the χ2 test to compare the rate of rare germline variants in complement genes in different groups. P < .05 was considered significant for all analyses. STATA v23 (STATA Corp) was used to perform analyses.
Results
Patient characteristics
From January 2015 through June 2019, we enrolled 59 patients with thrombotic APS, 74 patients with SLE, and 10 patients with CAPS (supplemental Table 1). Among the APS patients, 22 (37.3%) were single positive, 15 (25.4%) were double positive, and 22 (37.3%) were triple positive; 37 had had venous thrombosis, 13 had had arterial thrombosis, and 9 had had both venous and arterial thrombotic events. Recurrent thrombotic events occurred in 37.3% (22 of 59) of the APS patients, of which 14 developed on therapeutically dosed anticoagulation (including 3 while receiving a direct oral anticoagulant).
Thrombotic APS is associated with complement activation
Complement activity was assessed via complement-mediated killing of nucleated cells measured using the mHam assay, which has been validated in both disease states and normal subjects. The mHam assay (>20% cell killing)42,44,47 was positive in 35.6% (21 of 59) of patients with thrombotic APS and 85.7% (6 of 7 with available sera) of those with CAPS, compared with 6.8% (5 of 74) of those with SLE (P < .001; Figure 1A).
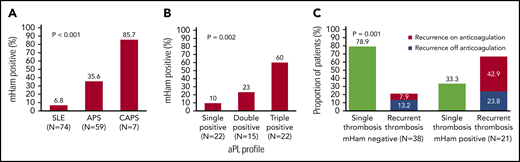
Complement activation in thrombotic APS and CAPS. (A) Complement activation indicated by a positive mHam assay was detected in 35.6% of patients with APS and 85.7% of those with CAPS, compared with only 6.8% patients with SLE (P < .001). The percentage of patients with a positive mHam assay also increased in a triple-positive aPL profile (positive for lupus anticoagulant and anti-β2GPI and anti-cardiolipin antibodies) (B) and recurrent thrombosis (C). P values refer to the χ2 test for trend in all cases.
Complement activation in thrombotic APS and CAPS. (A) Complement activation indicated by a positive mHam assay was detected in 35.6% of patients with APS and 85.7% of those with CAPS, compared with only 6.8% patients with SLE (P < .001). The percentage of patients with a positive mHam assay also increased in a triple-positive aPL profile (positive for lupus anticoagulant and anti-β2GPI and anti-cardiolipin antibodies) (B) and recurrent thrombosis (C). P values refer to the χ2 test for trend in all cases.
Among the patients with APS, mHam positivity was associated with triple positivity (60%), more than double (23%) or single positivity (10%) (P = .002; Figure 1B). mHam positivity was also more common with recurrent thrombosis (66.7% of patients with a positive mHam; 42.9% recurrence when receiving anticoagulation and 23.8% recurrence when off anticoagulation) than in those with a single event (33.3%). APS patients with a negative mHam assay were more likely to have had a single thrombotic event (78.9%) than recurrent thrombosis (21.1%; 7.9% recurrence when on anticoagulation and 13.2% recurrence when off anticoagulation; P = .001; Figure 1C). For patients with a known date of the most recent thrombosis, the mHam assay was positive in 68.4% (13 of 19) of patients when thrombosis occurred within 1 year before testing, compared with 31.8% (7 of 22) of patients with their most recent thrombotic event occurring >1 year before testing (P = .019; supplemental Figure 1). A positive mHam assay >1 year after thrombosis was associated with recurrent VTE (85.7% [6 of 7]; P = .014) and triple positivity (71.4% [5 of 7]; P = .04).
The mHam was positive in 43.8% (14 of 32) of patients with positive anti-β2GPI antibodies compared with 25.9% (7 of 27) of patients who had only aCL antibodies (P = .154). Among patients with anti-β2GPI antibodies, the mHam assay was positive in 50% (11 of 22) with anti-β2GPI IgG, 20% (1 of 5) with anti-β2GPI IgM, and 40% (2 of 5) with anti-β2GPI IgG and IgM (P = .300; supplemental Figure 2).
We evaluated deposition of terminal complement protein complexes (C5b-9) on PIGA− TF-1 cells after incubation with patient serum. APS patient sera induced C5b-9 deposition, which correlated with cell killing in the mHam assay (Figure 2A). In all cases, C5b-9 deposition was inhibited by blocking the terminal pathway of complement with an anti-C5 antibody. The factor D inhibitor ACH-4471 specifically blocks the alternative pathway.46 We previously demonstrated that ACH-4471 is highly effective in blocking complement-dependent killing in the mHam with aHUS serum (Figure 2B).46 However, ACH-4471 did not prevent cell killing in the mHam or prevent the C5b-9 deposition induced by APS sera to any appreciable extent, suggesting that complement activation in APS sera primarily occurs through the classical pathway. Consistent with this, C5b-9 staining was not increased on the PIGA− TF1 cell line when incubated with sera in alternative pathway–only buffer (GVB0- Mg-EGTA; data not shown) compared with a buffer that is not pathway selective (GVB++). Patients with a positive mHam result demonstrated significantly higher mean C5b-9 deposition than did the negative controls (29.2% vs 8.6%; P = .006), whereas patients with a negative mHam result did not show significantly increased mean C5b-9 deposition compared with heat inactivated negative controls (12.6% vs 6.5%, P = .331).
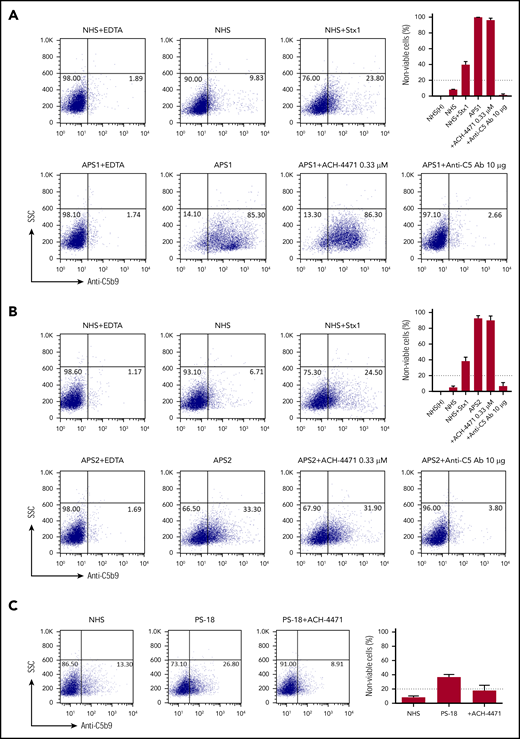
APS sera induce C5b-9 deposition. Flow cytometry demonstrated C5b-9 (membrane attack complex) deposition on the surface of PIGA− TF-1 cells in 2 representative patients: APS1 (A) and APS2 (B). Sera from both patients led to C5b-9 deposition, which was completely blocked in the presence of eculizumab (anti-C5 monoclonal antibody). Adding the factor D inhibitor ACH-4471 did not appreciably inhibit C5b-9 deposition in either patient, an observation that is also reflected in the mHam results shown in the top right of both panels. In the mHam, the dotted line at 20% nonviable cells indicates the threshold for a positive assay. (C) aHUS patient PS-18. ACH-4471 completely inhibited C5b-9 deposition induced by sera from the patient. Stx1, Shiga toxin 1 (positive control); SSC, side scatter; anti-C5 Ab, eculizumab; ACH-4471, factor D inhibitor.
APS sera induce C5b-9 deposition. Flow cytometry demonstrated C5b-9 (membrane attack complex) deposition on the surface of PIGA− TF-1 cells in 2 representative patients: APS1 (A) and APS2 (B). Sera from both patients led to C5b-9 deposition, which was completely blocked in the presence of eculizumab (anti-C5 monoclonal antibody). Adding the factor D inhibitor ACH-4471 did not appreciably inhibit C5b-9 deposition in either patient, an observation that is also reflected in the mHam results shown in the top right of both panels. In the mHam, the dotted line at 20% nonviable cells indicates the threshold for a positive assay. (C) aHUS patient PS-18. ACH-4471 completely inhibited C5b-9 deposition induced by sera from the patient. Stx1, Shiga toxin 1 (positive control); SSC, side scatter; anti-C5 Ab, eculizumab; ACH-4471, factor D inhibitor.
Patient-derived aPL activated complement in vitro
To further investigate whether aPL directly activates complement, we evaluated C5b-9 deposition on PIGA− TF1 cells incubated with serum to which affinity-purified, patient-specific anti-β2GPI antibodies were added (patient APS21, with thrombotic APS and triple-positive aPL profile including IgG anti-β2GPI, and patient APS3, with a positive aPL profile including IgM anti-β2GPI and no IgG anti-β2GPI, but no history of thrombosis). Anti-β2GPI antibodies from the patient with thrombotic APS induced C5b-9 deposition, which was inhibited by adding anti-C5 monoclonal antibody and minimally inhibited by ACH-4471 (Figure 3A). Anti-β2GPI antibodies from patient APS3 (with positive LA, IgM anti-β2GPI, and IgM aCL and no thrombotic events) led to a very small increase in C5b-9 deposition (Figure 3B), suggesting that complement activation is a characteristic of aPLs that are associated with thrombosis.
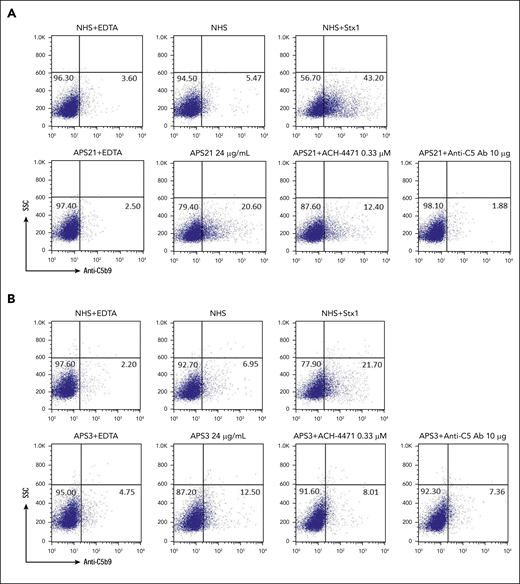
Pathogenic anti-β2GPI IgG induces C5b-9 deposition. (A) Patient APS21, anti-β2GPI IgG. Flow cytometry demonstrated that IgG anti-β2GPI antibody from patient APS21 with thrombotic APS induced C5b-9 (membrane attack complex) deposition on the surface of PIGA− cells. C5b-9 deposition was blocked completely with eculizumab and partially by the factor D inhibitor ACH-4471. Patient APS21 was a 30-year-old woman with venous thrombosis and recurrent pulmonary hemorrhage. aPL profile: LA+, aCL IgG+, aCL IgM−, anti-β2GPI IgG+, and anti-β2GPI IgM−. (B) Patient APS3, anti-β2 GP1 IgM. The IgM led to a very small increase in C5b-9 deposition. APS3 was a 51-year-old woman with persistently positive aPLs, but no thrombosis. aPL profile: LA+, aCL IgG−, aCL IgM+, anti-β2GPI IgG−, and anti-β2GPI IgM+. NHS(H), heat-inactivated NHS; Stx1, Shiga toxin 1 (positive control); SSC, side scatter; anti-C5 Ab, eculizumab; ACH-4471, factor D inhibitor.
Pathogenic anti-β2GPI IgG induces C5b-9 deposition. (A) Patient APS21, anti-β2GPI IgG. Flow cytometry demonstrated that IgG anti-β2GPI antibody from patient APS21 with thrombotic APS induced C5b-9 (membrane attack complex) deposition on the surface of PIGA− cells. C5b-9 deposition was blocked completely with eculizumab and partially by the factor D inhibitor ACH-4471. Patient APS21 was a 30-year-old woman with venous thrombosis and recurrent pulmonary hemorrhage. aPL profile: LA+, aCL IgG+, aCL IgM−, anti-β2GPI IgG+, and anti-β2GPI IgM−. (B) Patient APS3, anti-β2 GP1 IgM. The IgM led to a very small increase in C5b-9 deposition. APS3 was a 51-year-old woman with persistently positive aPLs, but no thrombosis. aPL profile: LA+, aCL IgG−, aCL IgM+, anti-β2GPI IgG−, and anti-β2GPI IgM+. NHS(H), heat-inactivated NHS; Stx1, Shiga toxin 1 (positive control); SSC, side scatter; anti-C5 Ab, eculizumab; ACH-4471, factor D inhibitor.
CAPS is associated with complement activation and rare germline variants in complement genes
We studied 10 patients with CAPS (Table 1). Acute phase sera were available for 7 patients and a positive mHam assay and increased C5b-9 deposition were found in 85.7% (6 of 7). The only patient with a negative mHam assay during acute CAPS had a sample obtained after 5 plasma exchanges had been completed. Similar to APS, CAPS patients with positive mHam results also demonstrated increased C5b-9 deposition on flow cytometry, which was blocked by eculizumab. C5b-9 deposition and mHam cell killing was partially blocked by ACH-4471 and was completely blocked by anti-C5 monoclonal antibody (representative example in Figure 4A).
Clinical characteristics, outcomes and complement studies in patients with CAPS
| Pt. No. | Age/sex | Presentation | Definite/Probable CAPS | Prior APS diagnosis | Trigger | aPL profile* | Repeat aPL profile* (at least 12 wk apart) | mHam+ | Mutations† |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 45/M | Ischemic toe after initiation of ITP therapy, DVT, pulmonary embolism, myocardial infarction, and renal insufficiency | Probable | + | ITP treatment (eltrombopag) | DRVVT+, hexagonal PL+, PNP+, aβ2GP-I IgG 52, aβ2GP-I IgM 34, aCL IgG 126, aCL IgM 24 | DRVVT+, hexagonal PL+, PNP+, aβ2GP-I IgG >150, aβ2GP-I IgM 52, aCL IgG >150, aCL IgM 38 | — | None |
| 2 | 37/F | Subdural hematoma, cardiomyopathy, acute kidney injury, and thrombocytopenia | Probable | + | Myocarditis, possibly viral | DRVVT+, hexagonal PL+, PNP+, aβ2GP-I >150, aβ2GP-I IgM 146, aCL IgG 127, aCL IgM 69 | DRVVT+, hexagonal PL+, PNP+, aβ2GP-I >107, aβ2GP-I IgM >150, aCL IgG 121, aCL IgM 120 | — | None |
| 3 | 40/M | Renal failure (biopsy showed cortical thrombosis and TMA), thrombocytopenia, and pulmonary embolism | Definite | + | Acute appendicitis | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | + | THBD |
| 4 | 67/F | Pulmonary emboli, arterial thrombosis requiring leg amputation (pathology showed widespread small-vessel thrombosis), digital gangrene, and renal failure | Probable | − | Rheumatoid arthritis flare | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | Repeat was not obtained | + | CFHR1- CFHR3, del (homozygous) |
| 5 | 34/F | Renal failure, thrombocytopenia and elevated liver enzymes | Probable | + | Pregnancy | DRVVT+, aCL IgG 131 | DRVVT+, aCL IgG 64 | + | CR1 |
| 6 | 63/M | Thrombocytopenia, renal failure, cardiomyopathy with reduced ejection fraction, adrenal hemorrhage, and skin ulcers. Renal biopsy showed TMA | Definite | + | Pyelonephritis | DRVVT+, aCL IgG <10, aCL IgM 28, aβ2GP-I IgG 31, aβ2GP-I IgM <10 | DRVVT+, aCL IgG <10, aCL IgM 28, aβ2GP-I IgG 41, aβ2GP-I IgM <10 | + | CFHR4, CR1 |
| 7 | 49/F | Arterial thrombosis of lower extremities, renal failure, cardiac ischemia, gangrene of the fingertips, thrombocytopenia, and skin necrosis | Probable | + | None | DRVVT+, aCL IgG 62, aCL IgM 66, aβ2GP-I IgG <10, aβ2GP-I IgM 40 | Not repeated after CAPS episode because patient died. Previous testing (>1 y prior) showed: DRVVT+, aCL IgG 21, aCL IgM 25, aβ2GP-I IgG 31, aβ2GP-I IgM 15 | − | CR1 |
| 8 | 50/M | Hepatic, spleen, and retinal infarction and renal failure | Probable | + | Acute cholecystitis and cholecystectomy | DRVVT+, aCL IgG >100, aCL IgM <10, aβ2GP-I IgG >100, aβ2GP-I IgM 14 | DRVVT+, aCL IgG, >100, aCL IgM <10, aβ2GP-I IgG >100, aβ2GP-I IgM 14 | + | None |
| 9 | 46/F | Diffuse hepatic infarction and evidence of microvascular thrombosis in the lungs, spleen, and possibly the kidneys. Relapsed about 6 mo after initial presentation with recurrent hepatic infarcts. | Probable | − | None | DRVVT+, aCL IgG 143, aCL IgM 73, aβ2GP-I IgG 61 | aCL IgG 52.5 (on plasmapheresis); DRVVT not repeated because of ongoing anticoagulation | + | CFHR1- CFHR3, del (homozygous) |
| 10 | 51/M | Multiple thrombi on mitral valve, renal failure, infarcts and ischemic injury of liver and spleen, and multiple small-vessel strokes | Definite | + | Mitral valve replacement | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | — | None |
| Pt. No. | Age/sex | Presentation | Definite/Probable CAPS | Prior APS diagnosis | Trigger | aPL profile* | Repeat aPL profile* (at least 12 wk apart) | mHam+ | Mutations† |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 45/M | Ischemic toe after initiation of ITP therapy, DVT, pulmonary embolism, myocardial infarction, and renal insufficiency | Probable | + | ITP treatment (eltrombopag) | DRVVT+, hexagonal PL+, PNP+, aβ2GP-I IgG 52, aβ2GP-I IgM 34, aCL IgG 126, aCL IgM 24 | DRVVT+, hexagonal PL+, PNP+, aβ2GP-I IgG >150, aβ2GP-I IgM 52, aCL IgG >150, aCL IgM 38 | — | None |
| 2 | 37/F | Subdural hematoma, cardiomyopathy, acute kidney injury, and thrombocytopenia | Probable | + | Myocarditis, possibly viral | DRVVT+, hexagonal PL+, PNP+, aβ2GP-I >150, aβ2GP-I IgM 146, aCL IgG 127, aCL IgM 69 | DRVVT+, hexagonal PL+, PNP+, aβ2GP-I >107, aβ2GP-I IgM >150, aCL IgG 121, aCL IgM 120 | — | None |
| 3 | 40/M | Renal failure (biopsy showed cortical thrombosis and TMA), thrombocytopenia, and pulmonary embolism | Definite | + | Acute appendicitis | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | + | THBD |
| 4 | 67/F | Pulmonary emboli, arterial thrombosis requiring leg amputation (pathology showed widespread small-vessel thrombosis), digital gangrene, and renal failure | Probable | − | Rheumatoid arthritis flare | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | Repeat was not obtained | + | CFHR1- CFHR3, del (homozygous) |
| 5 | 34/F | Renal failure, thrombocytopenia and elevated liver enzymes | Probable | + | Pregnancy | DRVVT+, aCL IgG 131 | DRVVT+, aCL IgG 64 | + | CR1 |
| 6 | 63/M | Thrombocytopenia, renal failure, cardiomyopathy with reduced ejection fraction, adrenal hemorrhage, and skin ulcers. Renal biopsy showed TMA | Definite | + | Pyelonephritis | DRVVT+, aCL IgG <10, aCL IgM 28, aβ2GP-I IgG 31, aβ2GP-I IgM <10 | DRVVT+, aCL IgG <10, aCL IgM 28, aβ2GP-I IgG 41, aβ2GP-I IgM <10 | + | CFHR4, CR1 |
| 7 | 49/F | Arterial thrombosis of lower extremities, renal failure, cardiac ischemia, gangrene of the fingertips, thrombocytopenia, and skin necrosis | Probable | + | None | DRVVT+, aCL IgG 62, aCL IgM 66, aβ2GP-I IgG <10, aβ2GP-I IgM 40 | Not repeated after CAPS episode because patient died. Previous testing (>1 y prior) showed: DRVVT+, aCL IgG 21, aCL IgM 25, aβ2GP-I IgG 31, aβ2GP-I IgM 15 | − | CR1 |
| 8 | 50/M | Hepatic, spleen, and retinal infarction and renal failure | Probable | + | Acute cholecystitis and cholecystectomy | DRVVT+, aCL IgG >100, aCL IgM <10, aβ2GP-I IgG >100, aβ2GP-I IgM 14 | DRVVT+, aCL IgG, >100, aCL IgM <10, aβ2GP-I IgG >100, aβ2GP-I IgM 14 | + | None |
| 9 | 46/F | Diffuse hepatic infarction and evidence of microvascular thrombosis in the lungs, spleen, and possibly the kidneys. Relapsed about 6 mo after initial presentation with recurrent hepatic infarcts. | Probable | − | None | DRVVT+, aCL IgG 143, aCL IgM 73, aβ2GP-I IgG 61 | aCL IgG 52.5 (on plasmapheresis); DRVVT not repeated because of ongoing anticoagulation | + | CFHR1- CFHR3, del (homozygous) |
| 10 | 51/M | Multiple thrombi on mitral valve, renal failure, infarcts and ischemic injury of liver and spleen, and multiple small-vessel strokes | Definite | + | Mitral valve replacement | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | DRVVT+, aCL IgG <10, aCL IgM <10, aβ2GP-I IgG <10, aβ2GP-I IgM <10 | — | None |
DRVVT, dilute Russell’s viper venom time; ITP, immune thrombocytopenia; DVT, deep venous thrombosis; hexagonal PL, hexagonal phospholipid; PNP, platelet neutralization procedure.
aCL IgG and IgM are expressed in IgG (GPL) and IgM phospholipid units (MPL), respectively. The threshold for a positive assay is 20 GPL for IgG aCL and 20 MPL for IgM. aβ2GP-I IgG and IgM are expressed in standard IgG units and standard IgM units, respectively. The threshold for a positive assay is 20 standard IgG units for IgG aβ2GP-I and 20 standard IgM units for IgM aβ2GP-I. A positive DRVVT indicates that the DRVVT confirm ratio (DRVVT/ DRVVT with added phospholipid) was >1.5.
Patients underwent targeted sequencing via a custom 15 gene panel. Only rare variants with minor allele frequency <0.005 in the genome aggregation database (gnomAD v2) were included.
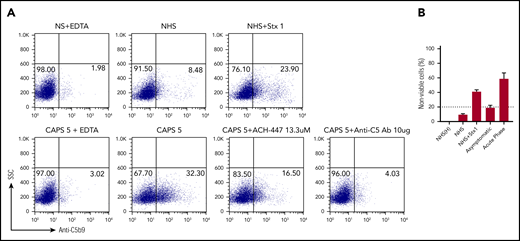
CAPS sera activate complement and induce C5b-9 deposition. (A) Sera from a representative patient with CAPS (CAPS5) induced C5b-9 deposition on PIGA− TF-1 cells. C5b-9 deposition was blocked by eculizumab (a C5 inhibitor) and partially by the factor D inhibitor ACH-4471. The mHam assay also showed a positive result (>20% nonviable cells) with CAPS sera, which was attenuated by eculizumab and a factor D inhibitor. (B) mHam was negative (>20% cell killing) when the patient was tested at steady state and positive when tested during acute CAPS. Patient CAPS5 had a CR1 V1675L variant; NHS, normal human serum; NHS(H), heat-inactivated NHS; Stx1, Shiga toxin 1 (positive control); SSC, side scatter; anti-C5 Ab, eculizumab; and ACH-4471, factor D inhibitor.
CAPS sera activate complement and induce C5b-9 deposition. (A) Sera from a representative patient with CAPS (CAPS5) induced C5b-9 deposition on PIGA− TF-1 cells. C5b-9 deposition was blocked by eculizumab (a C5 inhibitor) and partially by the factor D inhibitor ACH-4471. The mHam assay also showed a positive result (>20% nonviable cells) with CAPS sera, which was attenuated by eculizumab and a factor D inhibitor. (B) mHam was negative (>20% cell killing) when the patient was tested at steady state and positive when tested during acute CAPS. Patient CAPS5 had a CR1 V1675L variant; NHS, normal human serum; NHS(H), heat-inactivated NHS; Stx1, Shiga toxin 1 (positive control); SSC, side scatter; anti-C5 Ab, eculizumab; and ACH-4471, factor D inhibitor.
Serial samples were available for 2 patients with CAPS. The first patient was a 40-year-old man with a history of renal failure related to APS/CAPS, who developed acute renal failure, cardiac injury, and thrombocytopenia within 2 weeks of a renal transplant, which was successfully treated with eculizumab. The mHam assay was positive during this acute episode, but turned negative at 12 and 18 months after the acute episode. The second patient was a 36-year-old woman with triple-positive APS with recurrent venous thromboembolic events, despite anticoagulation, a history of hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome during pregnancy, as well as 2 prior episodes of CAPS in the postpartum setting. She was evaluated during her fourth pregnancy at 17 weeks of gestation (at which time she was asymptomatic) and the mHam assay was negative. However, a repeat mHam assay at 23 weeks of gestation was positive (Figure 4B). Two days after the second sample was drawn, she developed renal failure, thrombocytopenia, and elevated transaminases and was diagnosed with recurrent CAPS/HELLP. Clinical deterioration continued, even after emergent cesarean section. She was subsequently treated with plasma exchange and high-dose corticosteroids, in addition to anticoagulation, and ultimately recovered after a prolonged, complicated hospital course. The patient was not treated with eculizumab. Her severe thrombotic illness later in pregnancy may be explained by the fact that complement activity normally rises late in pregnancy48 and could have served as the trigger in this case.
Germline variants in genes critical for the function and regulation of the alternative pathway of complement contribute to complement-mediated diseases such as aHUS and HELLP.42,49 We hypothesized that patients with APS and CAPS harbor germline variants in complement genes and performed targeted sequencing of the exonic regions of 15 genes with a role in the alternative pathway of complement or previously implicated in the etiology of aHUS. Genomic DNA samples were available from 10 patients with CAPS, 55 patients with APS, and 21 patients with SLE (without aPL). To validate our custom sequencing panel, 33 patients with aHUS and 43 individuals without a diagnosis of a complement-mediated disorder were sequenced as positive and negative controls, respectively. Rare germline variants were present in 60.0% (6 of 10) patients with CAPS and 51.5% (17 of 33) of those with aHUS, compared with 21.8% (12 of 55) of patients with thrombotic APS, 28.6% (6 of 21) of those with SLE, and 23.3% (10 of 43) of unaffected individuals (Table 2). The frequency of germline variants in CAPS was significantly higher than in APS (P = .013), SLE (without aPL; P = .093), and controls (P = .022) and was similar to that in aHUS (P = .222). Rare variants in CAPS included (1) homozygous CFHR1-CFHR3 deletion (2 patients), (2) THBD P501L, (3) CR1 S1532G and homozygous CFHR1-CFHR3 deletion, (4) CFHR4 R287H and CR1 splice acceptor variant, and (5) CR1 V1675L (Figure 5; supplemental Table 2). A comprehensive list of all variants identified via targeted sequencing is presented in supplemental Table 3.
Summary of rare variants in complement genes
| Patients sequenced, n | Patients with rare variants, n* | Mutation rate (%) | |
|---|---|---|---|
| CAPS | 10 | 6 | 60.0 |
| APS | 55 | 12 | 21.8 |
| SLE | 21 | 6 | 28.6 |
| aHUS | 33 | 17 | 51.5 |
| Controls | 43 | 10 | 23.3 |
| Patients sequenced, n | Patients with rare variants, n* | Mutation rate (%) | |
|---|---|---|---|
| CAPS | 10 | 6 | 60.0 |
| APS | 55 | 12 | 21.8 |
| SLE | 21 | 6 | 28.6 |
| aHUS | 33 | 17 | 51.5 |
| Controls | 43 | 10 | 23.3 |
Genes included on the panel: CFH, CFI, CFB, CFD, CFP, CFHR1, CFHR2, CFHR3, CFHR4, CFHR5, CR1, C3, THBD, CD46, DGKE.
Defined as minor allele frequency <.005
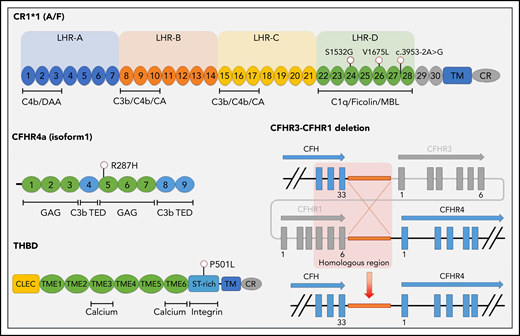
Germline variants identified in patients with CAPS. The single-nucleotide variant location is provided, along with a schematic diagram of the CFHR3-CFHR1 deletion (identified in 3 patients). Exons are indicated as blocks. Short consensus repeats (SCRs) are numbered for both CR1 (C3b/C4b receptor) and CFHR4, with the most common isoforms shown. Binding sites for calcium, integrins, and complement proteins are also shown. Protein domains include LHR, long homologous repeat; TM, transmembrane domain; CR, cytoplasmic region; CLEC, C-type lectin domain; TIME, thrombomodulin EGF-like domain. Regulatory regions include SA, sialic acids; C3b thioester-containing domain (TED); MBL, mannose binding lectin; CA, cofactor activity; DAA, decayed accelerating activity; GAG, glycosaminoglycan.
Germline variants identified in patients with CAPS. The single-nucleotide variant location is provided, along with a schematic diagram of the CFHR3-CFHR1 deletion (identified in 3 patients). Exons are indicated as blocks. Short consensus repeats (SCRs) are numbered for both CR1 (C3b/C4b receptor) and CFHR4, with the most common isoforms shown. Binding sites for calcium, integrins, and complement proteins are also shown. Protein domains include LHR, long homologous repeat; TM, transmembrane domain; CR, cytoplasmic region; CLEC, C-type lectin domain; TIME, thrombomodulin EGF-like domain. Regulatory regions include SA, sialic acids; C3b thioester-containing domain (TED); MBL, mannose binding lectin; CA, cofactor activity; DAA, decayed accelerating activity; GAG, glycosaminoglycan.
Discussion
APS serum demonstrated complement activation shown by a functional assay (mHam) and increased C5b-9 deposition on the cell surface, which was recapitulated on adding patient-derived anti-β2GPI antibodies to normal sera. CAPS patients have a high rate of rare germline variants in complement regulatory genes, which may serve as a second hit (in addition to aPL), leading to uncontrolled complement activation and a more severe clinical phenotype.
Although the aPL profile helps in risk stratifying of patients with APS,21 the absence of a reliable biomarker to distinguish clinically relevant from clinically irrelevant aPL (and to predict which patients will have first or recurrent thrombosis) is a major gap in APS clinical care. Previous studies have demonstrated hypocomplementemia36 and increased levels of complement activation byproducts including fragment Bb, C3a, and C5b-9 in patients with APS,33-35 but have not consistently shown an association with thrombosis.35,36 Serum levels of complement byproducts measure complement activation indirectly, and it is difficult to establish clinically relevant thresholds on these assays. We used a functional assay to show that complement activation is associated with patients with aPL who had thrombosis, and patients with a more severe APS phenotype characterized by recurrent thrombosis are more likely to have evidence of persistent complement activation. Of the patients with thrombotic APS, ∼35% had a positive mHam assay compared with 6.8% of controls with SLE. The mHam result was positive in 68.4% of patients enrolled within a year of their thrombotic events compared with 31.8% of patients enrolled beyond 1 year. Most patients with positive mHam assays beyond a year after their thrombotic event had had recurrent thrombosis and a triple-positive aPL profile. Our findings suggest that complement activation, as measured in our assay, may be a marker of more clinically important APS. The mHam assay was positive in a higher proportion of patients with anti-β2GPI aPLs compared with patients with other aPLs (43.8% vs 25.9%; P = .154). In addition, the mHam was more likely to be positive in patients with IgG anti-β2GPI (50%) or both IgG and IgM anti-β2GPI (40%) than in those with IgM anti-β2GPI alone (20%; P = .467). However, these differences were not statistically significant, most likely because of the small number of patients in these subgroups. This trend is consistent with the stronger association of IgG aPLs with thrombotic events.50
Anti-β2GPI IgG from patients with thrombotic APS lead to membrane attack complex (C5b-9) deposition on the cell surface, which echoes murine models of APS in which aPL-induced deposition of complement proteins on the vascular endothelium and thrombosis were attenuated in C6-deficient (C6−/−) rats or animals treated with a C5 inhibitor.16 Meroni et al28 also showed that C5b-9 colocalized with β2GP-I and IgG in the arterial wall of a patient with APS and recurrent arterial thrombosis who responded to treatment with eculizumab. In contrast, anti-β2GPI IgM from a patient without thrombosis did not activate complement, highlighting that complement activation is a critical characteristic of pathogenic aPLs. Agostinis et al17 showed that a non–complement-fixing version of an anti-β2GPI antibody lost the ability to induce thrombosis in a rat model. The link between complement activation and thrombosis is illustrated by experiments showing that aPL-induced complement activation leads to tissue-factor–dependent procoagulant activity related to the effects of C5a on neutrophils,51 monocytes,52 and endothelial cells,53 as well endothelial tissue factor expression induced by C5b-9.54
The mechanisms by which anti-β2GPI antibodies cause complement activation remain unclear; however, mechanisms involving autoantibodies against factor H,55-57 a regulatory role of cell-surface–bound β2GP-I,58 and activation of the classical complement pathway through C1q have been proposed.59 We found that complement activation (cell killing and C5b-9 deposition) in APS sera or induced by patient-derived aPLs was blocked by a terminal complement inhibitor (anti-C5 antibody) and less so by an alternative pathway selective inhibitor (ACH-4471) suggesting that aPL induces complement activation primarily through the classical complement pathway. In contrast, complement-dependent cell killing and C5b-9 deposition induced by CAPS sera were partially inhibited by ACH-4471 (and more so by anti-C5 antibody) suggesting activation of both the classic and alternative pathways of complement. This finding may be explained by the higher rate of mutations in genes regulating the alternative complement pathway in patients with CAPS.
Rare germline variants in complement genes were detected in 60% of patients with CAPS. Acute CAPS was also characterized by complement activation highlighting the clinical and pathogenic similarities between CAPS and aHUS, the prototypical complement-mediated microangiopathy.60 Our group recently demonstrated complement activation and germline complement gene variants in patients with the HELLP syndrome,42 which is several-fold more common in patients with APS and exhibits marked clinical overlap with CAPS.61 We propose a pathogenic model in which aPLs are the first hit and can induce complement activation and cause thrombosis, whereas patients who also have a pathogenic complement regulatory gene mutation (second hit) are predisposed to uncontrolled complement activation, leading to CAPS in the setting of a complement-amplifying trigger, such as infection, surgery, pregnancy, or autoimmune disease (Figure 6).62 Several reports describe the successful use of eculizumab to treat CAPS29-32 and thrombotic APS refractory to anticoagulation28 ; however, mechanistic data and disease biomarkers have been lacking. Our findings provide a rationale for studying complement inhibition as a therapeutic strategy for CAPS and anticoagulation-refractory APS.
![Proposed 2-hit model for CAPS. We propose a pathogenic model in which aPLs induce complement activation and cause thrombosis. Patients who also have a pathogenic loss-of-function mutation in a complement inhibitory factor (CFH, CFI, CD46 [MCP], THBD, CR1) or a gain-of-function mutation of a complement-activating factor (CFB, C3) are likely to be predisposed to uncontrolled complement activation, which could lead to disseminated thrombosis and ischemic multiorgan failure in the setting of a complement-amplifying trigger, such as infection, surgery, or autoimmune disease.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/4/10.1182_blood.2019003863/5/m_bloodbld2019003863f6.png?Expires=1769134533&Signature=LKeREy2Ysgw2XsaciLeBqCO0cj9NN-KcAWjqFL3Rqj4xiF-jtRnxZ9RbTzbWPVVvukFFy1y2bjmw3KUE5rvl~7GypTaZVTdlEl6dq00PnMLWPqyag7Wh5SJbb69xqIz56bv28A36FpMyMKm8mVhnJ6S5lqB1vBXCsQWKvgCKUgGwmYeoQYIQf7XqAIsLuZPmuQxvK7aXa~KnO~K3t6YqKhHL-t7IMVn7cFW0ULlcEInCz4Zb0gd7Xeim43pAeH5YJfAZB7RXewS1Zh4th65Mzy-JsvkDUEBoaFHFoYnBL6w8NQKy3BYfXILoVVPN3XZZeMdQ1RTBI4IGKf3vo4th9w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Proposed 2-hit model for CAPS. We propose a pathogenic model in which aPLs induce complement activation and cause thrombosis. Patients who also have a pathogenic loss-of-function mutation in a complement inhibitory factor (CFH, CFI, CD46 [MCP], THBD, CR1) or a gain-of-function mutation of a complement-activating factor (CFB, C3) are likely to be predisposed to uncontrolled complement activation, which could lead to disseminated thrombosis and ischemic multiorgan failure in the setting of a complement-amplifying trigger, such as infection, surgery, or autoimmune disease.
Proposed 2-hit model for CAPS. We propose a pathogenic model in which aPLs induce complement activation and cause thrombosis. Patients who also have a pathogenic loss-of-function mutation in a complement inhibitory factor (CFH, CFI, CD46 [MCP], THBD, CR1) or a gain-of-function mutation of a complement-activating factor (CFB, C3) are likely to be predisposed to uncontrolled complement activation, which could lead to disseminated thrombosis and ischemic multiorgan failure in the setting of a complement-amplifying trigger, such as infection, surgery, or autoimmune disease.
Limitations of our study include that we evaluated a highly selected group of patients who were referred to tertiary care centers and may not be fully representative of all APS patients. Patients were not recruited consecutively and were not matched to the control group (SLE) for treatments or comorbidities. Time from event to sampling was variable, and we did not have serial samples from enough patients to draw robust conclusions regarding the persistence of complement activation over time. Finally, we do not currently have functional data other than the mHam result to confirm the pathogenic significance of these germline variants; however, their frequency is comparable to that found in patients with aHUS60 and significantly higher than in control groups without TMA.
In summary, anti-β2GPI antibodies from patients with APS activate complement, and complement activation correlates with thrombotic events in APS. Moreover, most patients with CAPS have underlying complement regulatory gene variants that predispose to increased complement activity and a fulminant course with widespread thrombosis and multiorgan failure.
Please contact the corresponding author at brodsro@jhmi.edu for original data pertaining to this manuscript. No identifiable data will be shared. Requests for de-identified data from internal or external investigators will be evaluated on an individual basis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health (NIH), Heart, Lung and Blood Institute grants R01HL133113 (R.A.B), R01HL123098 (K.R.M.), and K08HL138142 (E.M.B.); NIH, National Institute of Arthritis and Musculoskeletal and Skin Diseases grant R01AR069572 (M.P.); and a Hemostasis and Thrombosis Research Society (HTRS) Mentored Research Award (S.C.).
Authorship
Contribution: S.C. collected and managed the data, enrolled patients, designed and performed the analyses, interpreted the data, and wrote the first draft of the manuscript; E.M.B. enrolled patients, designed and performed the genetic sequencing and analysis and interpreted the results, and edited the manuscript; X.Y., A.A., J.Y., and E.G. designed and performed the experiments and interpreted the data; H.C. performed the genetic sequencing and analysis; R.A. prepared patient-derived aPLs; M.B.S. and M.P. enrolled patients; M.A.C. enrolled patients; K.R.M. provided purified patient-derived aPL, enrolled patients, and interpreted the results; R.A.B. designed the study, supervised the experiments, interpreted the data, and wrote portions of the manuscript; and all authors critically reviewed the manuscript and approved the final version.
Conflict-of-interest disclosure: M.A.C. has served on data safety monitoring boards supported by Bayer and on advisory bodies for Servier Canada and Asahi Kasei; has prepared educational materials and/or presented such materials for Pfizer, CSL Behring, and Diagnostica Stago; and owns stock in Alnylam. S.C. has served on advisory boards for Alexion and Sanofi. R.A.B. has served on the scientific advisory boards of Alexion and Achillion. The remaining authors declare no competing financial interests.
Correspondence: Robert A. Brodsky, Division of Hematology, Johns Hopkins University School of Medicine, Ross Research Building, Room 1025, 720 Rutland Ave, Baltimore, MD 21205-2196; e-mail: brodsro@jhmi.edu.
