

Key Points
F erythroblasts are highly similar to non-HbF–expressing cells in global transcriptome, proteome, and expression of known HbF regulators.

Abstract
Reversing the developmental switch from fetal hemoglobin (HbF, α2γ2) to adult hemoglobin (HbA, α2β2) is an important therapeutic approach in sickle cell disease (SCD) and β-thalassemia. In healthy individuals, SCD patients, and patients treated with pharmacologic HbF inducers, HbF is present only in a subset of red blood cells known as F cells. Despite more than 50 years of observations, the cause for this heterocellular HbF expression pattern, even among genetically identical cells, remains unknown. Adult F cells might represent a reversion of a given cell to a fetal-like epigenetic and transcriptional state. Alternatively, isolated transcriptional or posttranscriptional events at the γ-globin genes might underlie heterocellularity. Here, we set out to understand the heterogeneity of HbF activation by developing techniques to purify and profile differentiation stage-matched late erythroblast F cells and non–F cells (A cells) from the human HUDEP2 erythroid cell line and primary human erythroid cultures. Transcriptional and proteomic profiling of these cells demonstrated very few differences between F and A cells at the RNA level either under baseline conditions or after treatment with HbF inducers hydroxyurea or pomalidomide. Surprisingly, we did not find differences in expression of any known HbF regulators, including BCL11A or LRF, that would account for HbF activation. Our analysis shows that F erythroblasts are not significantly different from non-HbF–expressing cells and that the primary differences likely occur at the transcriptional level at the β-globin locus.
Introduction
Reactivating the expression of fetal hemoglobin (HbF, α2γ2) in adult erythroblasts is an important goal in the treatment of sickle cell disease (SCD) and β-thalassemia. HbF can be found in small amounts in healthy individuals as well as at somewhat elevated levels in patients with SCD or other disorders of hematopoiesis such as Diamond-Blackfan anemia.1 HbF is generally found distributed in a heterocellular manner in a subset of cells termed “F cells.” Surprisingly, treatment with pharmacologic HbF inducers, such as hydroxyurea, or some non-deletional genetic lesions in γ-globin gene regulatory elements that lead to hereditary persistence of fetal hemoglobin, still result in a heterocellular HbF distribution despite affecting all erythroid progenitors.2-4 Therapeutic approaches to increase HbF expression in SCD currently aim to increase the total number of F cells that contain a threshold amount of HbF to counteract sickling.5
The presence of a distinct population of HbF-expressing cells was first noted in transfused patients and biochemical studies in the 1950s,6,7 and subsequently observed in blood smears using Hb solubility differences.8,9 The HbF-containing population in SCD patients showed improved cell survival and reduced sickling.6,7,10 Improved imaging techniques and antibodies in the 1970s allowed better characterization of F cells in healthy individuals and under conditions of erythroid stress; they also helped establish that F cells still contain predominantly adult hemoglobin (HbA) and that increases in total HbF by high performance liquid chromatography correlate with the number of F cells.11,12 One potential explanation for a distinct F-cell population is the persistence of fetal-like stem or progenitor cells in adults. Arguing against this are studies of clonal hematopoiesis in polycythemia vera and paroxysmal nocturnal hemoglobinuria,13,14 as well as in vitro studies of single-cell derived burst-forming unit erythroid colonies that contain F and non-F cells.15-17 These experiments suggest that F cells likely arise instead from erythroid precursors through a stochastic process with different probabilities of F-cell formation along the spectrum of erythroid development.13-19 Recent experiments with cultures starting with single-cell common myeloid progenitors and their daughter cells confirmed the earlier findings.20
One hypothesis for heterogeneous HbF expression posits that a fraction of adult erythroid progenitors reverts to a fetal-like state. Profiling of fetal liver– and adult bone marrow–derived erythroid cultures by several groups showed large numbers of differentially expressed transcripts (ranging from 600 to 3129) associated with stage-specific enhancers.21-24 Another hypothesis is that HbF heterogeneity is caused by much more limited changes in gene expression, selectively involving only 1 or a few of the known HbF regulators or their co-regulators. BCL11A and LRF/ZBTB7A, along with their corepressors (eg, nucleosome remodeling and deacetylase [NuRD] complex), are critical for silencing the γ-globin genes,25-30 and additional regulators were recently characterized.31-35 F cells could thus arise from precursors that variably, perhaps stochastically, express 1 or more of these regulators above or below a certain threshold needed to maintain HbF repression. For instance, a recent study of single-cell cultures showed variation in expression of BCL11A, KLF1, and TAL1 that correlated with response to hydroxyurea.20 Finally, the heterogeneity in HbF expression could be rooted solely in the β-globin locus itself. However, to date, none of these explanations for the origins of F cells have been directly tested. Studies of pharmacologic HbF induction have also not been able to answer the important question of why only a subset of cells is able to turn on HbF in response to treatment.
Despite more than 50 years of observations of F cells, it remains unclear why HbF production is restricted to only a subset of a genetically identical population at baseline conditions and in disease states or with pharmacologic treatment. This is largely a result of the technical challenges of analyzing F cells, since their purification via cell sorting requires fixation and permeabilization steps that are unsuitable for advanced downstream analysis of RNA and protein. We have optimized an approach that allows reversible fixation, sorting stage-matched HbF-positive or -negative populations, followed by crosslink reversal to obtain high-quality RNA and protein from cell lines and primary erythroid cultures. By using these newly developed techniques, we carried out the first direct in-depth analysis of carefully stage-matched F and A erythroblasts under various conditions.
Methods
Cells and cell culture
HUDEP2 and HUDEP2C cells stably expressing the CAS9 protein and culture conditions have been previously described.32,36 Single-cell–derived HUDEP2C clones were generated by limiting dilution. Clones that spontaneously resulted in high numbers of F cells (10% to 20%) on differentiation were then subcloned before HUDEP2 sorting experiments (see supplemental Methods, available online on the Blood Web site). CD34+ cells from peripheral blood of healthy donors were obtained from the Fred Hutchinson Cooperative Center for Excellence in Hematology (Seattle, WA) and cultured in a 3-phase medium as previously described.32 For pharmacologic treatment experiments, 50 µM hydroxyurea or 1 µM pomalidomide were added starting at day 6 with continued treatment until analysis.
Cell fixation, permeabilization, staining, and sorting
We optimized a previously published method for fixation and permeabilization of cells for subsequent RNA recovery.37 Cells were washed with phosphate-buffered saline, stained with Live/Dead fixable dead cell stain (Thermo Fisher Scientific, Waltham, MA), fixed for 30 minutes at 4°C in 2% to 4% formaldehyde, and permeabilized in 0.1% Triton X-100 for 5 minutes, then stained for HbF and cell surface markers CD71 and CD235. We tested multiple γ-globin antibodies in comparison with the current standard HbF allophycocyanin antibody from Thermo Fisher Scientific (MHFH05) and ultimately chose a sheep polyclonal antibody (Novus Biologicals, Centennial, CO) conjugated to AlexaFluor647, allowing for better separation of HbF cells at a significantly lower cost. Cells were sorted on a BD FACSAria Fusion Sorter for matched CD71+CD235+ fluorescence-activated cell sorter–matched populations, isolating either HbF+ or the bottom 20% of HbF– cells. We used RNase-free reagents throughout and included a recombinant RNase inhibitor or protease inhibitor for all steps. Additional details are available in the supplemental Methods.
RNA extraction, RT-PCR, and RNA-seq
RNA from sorted cells was purified using the RecoverAll Total Nucleic Acid Isolation Kit for formalin-fixed paraffin-embedded (FFPE) cells (Thermo Fisher Scientific) following the supplied protocol, except crosslink reversal was done at 50°C for 45 minutes. After RNA elution, a second DNase treatment step was performed using the DNA-free DNA Removal Kit (Thermo Fisher Scientific). Complementary DNAs were prepared by reverse transcription using iScript Supermix (Bio-Rad). Quantitative real-time polymerase chain reaction (qRT-PCR) using standard-curve quantification was performed with TaqMan Gene Expression Master Mix (Thermo Fisher Scientific) and TaqMan gene expression assays described in the supplemental Data and normalized to average expression of 3 housekeeping genes (ACTB, HPRT, and RPS18). RNA sequencing (RNA-seq) library preparation, sequencing using Illumina NextSeq 500, and processing were performed as described in supplemental Methods after depletion of ribosomal RNA and globin RNA. RNA-seq data were analyzed using the DESeq2 package in R (R Foundation for Statistical Computing, Vienna, Austria), and gene set enrichment analysis (GSEA) was performed using standard methods.38,39 All RNA-seq data are deposited in the Gene Expression Omnibus (accession #GSE144052).
Protein analysis
Protein extraction and crosslink reversal were performed using the Qproteome FFPE Tissue Kit (Qiagen, Germantown, MD).40 For HUDEP2 cells, F and A cells from 3 subclones were sorted on day 6 of differentiation and purified protein size-fractionated into 8 fractions by polyacrylamide gel electrophoresis. Mass spectrometry sample preparation and analysis were performed by the Proteomics and Metabolomics Facility at the Wistar Institute (Philadelphia, PA). Tryptic digests were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a Thermo Q Exactive Plus Mass Spectrometer (Thermo Fisher Scientific). MS/MS data were searched against the UniProt human database using MaxQuant 1.6.2.3 and analyzed as described in supplemental Methods. Fluorescent western blotting was performed using standard protocols with the antibodies listed in supplemental Methods.
Results
A protocol for F-erythroblast purification that allows transcriptome and proteome studies
Previous studies of F cells have been encumbered because most fixation protocols for erythroid cells do not sufficiently preserve nucleic acids for downstream analysis. However, recent interest in analysis of specific populations of fixed cells or FFPE samples has led to new approaches for recovering RNA and protein.37,40 We optimized a fixation protocol using 2% to 4% formaldehyde and permeabilization with 0.1% Triton-X100 that allows staining for both intracellular HbF and cell surface differentiation markers CD71 and CD235 that is otherwise similar to current glutaraldehyde fixation protocols. Formaldehyde fixation might potentially be more compatible with other methods such as chromatin immunoprecipitation sequencing and chromatin conformation capture. Using RNase and protease inhibitors throughout sample preparation and sorting produced RNA with RNA integrity numbers >9, similar to RNA isolated from non-fixed cells, and allowed for recovery of high-quality proteins. Finally, we tested multiple commercially available antibodies to γ-globin for hemoglobin chain specificity and reactivity in fixed cells, which identified a high-specificity sheep polyclonal antibody that allowed labeling of hundreds of millions of cells at low cost.
We validated our sorting approach by using HUDEP2 cells, a human erythroid cell line that recapitulates adult erythropoiesis and hemoglobin production and has been engineered by us to stably express spCas9 (HUDEP2C).32,36 Parental HUDEP2 cells are not clonal, and sorting for HbF+ cells may select particular clones within a population. To address this, we generated single-cell–derived HUDEP2C clones. Still, high clonal variability in HbF expression was found at baseline and upon pharmacologic HbF induction (supplemental Figure 1A-B), with no accompanying changes in BCL11A and LRF (supplemental Figure 1C-D). After differentiating 3 subclones, we sorted HbF+ (F) and HbF– (A) cells matched for CD71, CD235, and forward scatter (Figure 1A). qRT-PCR (Figure 1B) showed ∼100-fold enrichment of HBG transcripts in F cells, with similar levels of HBA and HBB expression. Similarly, the fetal-specific long noncoding RNA BGLT3, coded within the β-globin locus, is also enriched in F cells.33 Other transcripts that reflect erythroid differentiation, such as AHSP, CA1, and KLF1 were unchanged between F and A cells, validating our stage-matching approach. Western blot analysis was concordant with the RNA results (Figure 1C). Droplet digital RT-PCR (ddRT-PCR), which more accurately measures globin transcripts in sorted populations (Figure 1D), suggests that γ-globin accounts for ∼30% of total globin transcripts, similar to 14% to 28% previously noted by quantitative immunofluorescence in adult erythrocyte F cells.11
![Sorting stage-matched F and Aerythroblasts. (A) Sorting scheme for HUDEP2 and CD34+ hematopoietic stem and progenitor cell (HSPC)-derived cells. Cells are gated using a viability marker and then matched for forward scatter, CD71, and CD235 expression. After sorting, cells generally have ∼95% to 99% purity. (B) RT-PCR validation of HUDEP2 F and A cell sorts from 3 HUDEP2C clones. Data are normalized to the average of 3 housekeeping genes and shown relative to A cells. (C) Western blot validation with sorted cells from 3 HUDEP2 clones. (D) ddRT-PCR analysis of globin transcripts in sorted F and A cells, normalized to AHSP transcripts. Percentage of HBG transcripts (HBG/[HBG + HBD + HBB] × 100) is also shown. All data are shown as individual points, mean, and standard deviation (SD) for 3 samples. ***P < .001; **P < .01. NS, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/22/10.1182_blood.2020005058/4/m_bloodbld2020005058f1.png?Expires=1769096568&Signature=AjhjJth3fSq1c6zM~FP5cMK1zkCaHHf4ed65xBVPR~DYWFrbzs-goSg1Q7Mz4mh9XdxklAcCPe5pB~Qf5yBipEuN1nyfJgQnhx1wuyZbNHHpPkdlAWA3oBA3dGKjcX2FNRyORMSzFjfwWVefDBAYpgRvHZBptT-75ZMUoaOihR24kpAxxR4B7CG1JOZO7R9KThyJNPajIL7Ov9wSYUPjk1HuNgW7KwXLSFUpl0m0~fELSeuP7GI9gYxKpHQCvpkaPX9YoRqAFWRxReAgthYeFKcO1z~780RiHIbBQIZPe33lgJUl3q2OUM5MfYeGjuo7d~yVZHjCiozjHrsbBv6UNg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Sorting stage-matched F and Aerythroblasts. (A) Sorting scheme for HUDEP2 and CD34+ hematopoietic stem and progenitor cell (HSPC)-derived cells. Cells are gated using a viability marker and then matched for forward scatter, CD71, and CD235 expression. After sorting, cells generally have ∼95% to 99% purity. (B) RT-PCR validation of HUDEP2 F and A cell sorts from 3 HUDEP2C clones. Data are normalized to the average of 3 housekeeping genes and shown relative to A cells. (C) Western blot validation with sorted cells from 3 HUDEP2 clones. (D) ddRT-PCR analysis of globin transcripts in sorted F and A cells, normalized to AHSP transcripts. Percentage of HBG transcripts (HBG/[HBG + HBD + HBB] × 100) is also shown. All data are shown as individual points, mean, and standard deviation (SD) for 3 samples. ***P < .001; **P < .01. NS, not significant.
Sorting stage-matched F and Aerythroblasts. (A) Sorting scheme for HUDEP2 and CD34+ hematopoietic stem and progenitor cell (HSPC)-derived cells. Cells are gated using a viability marker and then matched for forward scatter, CD71, and CD235 expression. After sorting, cells generally have ∼95% to 99% purity. (B) RT-PCR validation of HUDEP2 F and A cell sorts from 3 HUDEP2C clones. Data are normalized to the average of 3 housekeeping genes and shown relative to A cells. (C) Western blot validation with sorted cells from 3 HUDEP2 clones. (D) ddRT-PCR analysis of globin transcripts in sorted F and A cells, normalized to AHSP transcripts. Percentage of HBG transcripts (HBG/[HBG + HBD + HBB] × 100) is also shown. All data are shown as individual points, mean, and standard deviation (SD) for 3 samples. ***P < .001; **P < .01. NS, not significant.
F-cell analysis in HUDEP2
We first analyzed sorted matched F and A cells in 3 HUDEP2C clones from the validation experiments. RNA-seq and proteomic analysis of erythroid cells can have impaired sensitivity because of high levels of globin transcripts and proteins. To improve detection of low abundance transcripts, we depleted abundant globin transcripts and ribosomal RNAs during generation of RNA-seq libraries, which reduced transcripts per million values for globin transcripts by about 1000-fold but still allowed for their detection. Similarly, for MS, we size-fractionated proteins to allow detection of approximately 6100 proteins, similar to reports from freshly isolated primary erythroblasts.41 We used stage-matched cells sorted on differentiation day 7 for RNA-seq (Figure 2A) and day 6 cells for MS (Figure 2B). Using a threshold of 1.5-fold change and an adjusted value of P < .05, the total number of differentially expressed genes was overall quite low, with 36 A-cell enriched transcripts and 58 F-cell enriched transcripts. The top enriched transcripts were HBG1, HBG2 (noting that globin messenger RNA [mRNA] depletion is incomplete and provides internal validation of F- vs A-cell enrichment), and BGLT3. Similarly, MS showed only a few differences between F and A cells. Of 6120 proteins that were detected in at least 1 of the sample groups, 19 were enriched in F cells and 5 were enriched in A cells using a threshold of greater than twofold change and Student t test value of P < .05. Using a more stringent false discovery rate–based statistical analysis, only 3 proteins were differentially enriched: γG-globin, γA-globin, and an immunoglobulin chain of the anti-HbF antibody. Only HBG1 and HBG2 were differentially expressed at both the RNA and protein levels. This remarkable congruence in transcriptome and proteome among F and A cells points to an unexpectedly high degree of similarity between these populations.
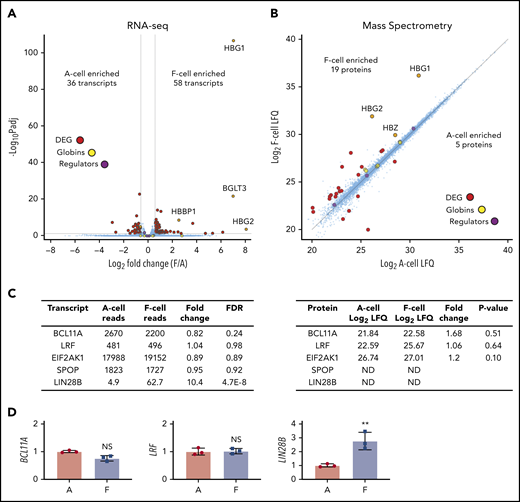
Analysis of HUDEP2-derived F and Aerythroblasts. (A) RNA-seq analysis of day 7 differentiated HUDEP2 F and A cells from 3 independent subclones. False discovery rate (FDR) <0.05; fold change >1.5. (B) MS analysis of day 6 differentiated HUDEP2 F and A cells. Approximately 6000 proteins were detected. P < .05; fold change >2. Globin transcripts and known HbF regulators (BCL11A, LRF, EIF2AK1, and SPOP) are highlighted. (C) Transcript (left) and protein (right) levels of selected HbF regulators. Transcript data are shown as normalized reads. (D) RT-PCR quantification of selected HbF regulators from sorted HUDEP2 day 7 F and A cells. Data are shown as mean ± SD. **P < .01 vs A cells. DEG, differentially expressed genes; LFQ, label-free quantification intensity; Padj, adjusted P value.
Analysis of HUDEP2-derived F and Aerythroblasts. (A) RNA-seq analysis of day 7 differentiated HUDEP2 F and A cells from 3 independent subclones. False discovery rate (FDR) <0.05; fold change >1.5. (B) MS analysis of day 6 differentiated HUDEP2 F and A cells. Approximately 6000 proteins were detected. P < .05; fold change >2. Globin transcripts and known HbF regulators (BCL11A, LRF, EIF2AK1, and SPOP) are highlighted. (C) Transcript (left) and protein (right) levels of selected HbF regulators. Transcript data are shown as normalized reads. (D) RT-PCR quantification of selected HbF regulators from sorted HUDEP2 day 7 F and A cells. Data are shown as mean ± SD. **P < .01 vs A cells. DEG, differentially expressed genes; LFQ, label-free quantification intensity; Padj, adjusted P value.
We anticipated changes in the levels of 1 or more of the known HbF regulators in F cells compared with A cells. Surprisingly, the well-established regulators BCL11A and LRF and the more recently characterized regulators HRI and SPOP were similar at both the transcript and protein levels (Figure 2C). The same held true for levels of NuRD components known to function as coregulators for BCL11A and LRF,27,29,30 as well as other associated epigenetic regulators (supplemental Tables 1 and 2). To ensure that we studied functionally relevant maturation stages, we also sorted cells derived from 3 independent HUDEP2C clones at several intervals starting at day 4 of differentiation, the earliest time at which we could reliably purify F cells (supplemental Figure 2A). Neither BCL11A nor LRF transcripts were significantly different at all time points examined.
Notably, we did detect elevated transcript levels of LIN28B in HUDEP2 F cells, but LIN28B protein escaped detection by MS and western blotting. LIN28B is an RNA binding protein that is expressed in a variety of tissues, including fetal hematopoietic cells, and it regulates stability and translation of multiple RNAs either directly or through the let-7 family of microRNAs.42-48 We confirmed increased LIN28B expression in F cells by qRT-PCR (Figure 2D; supplemental Figure 2A). Higher LIN28B transcript levels in F cells were intriguing because ectopic expression of LIN28B in various adult tissues can lead to reversion to a fetal-like phenotype42,45 ; in primary erythroblasts, LIN28B overexpression increases HbF production through decreasing BCL11A transcripts and protein.44 However, we did not find differences in LIN28B downstream targets HMGA2 and IGF2BP1 (supplemental Tables 1 and 2) or the LIN28B-repressed let-7 microRNAs (supplemental Figure 2B) between F and A erythroblasts. To determine whether LIN28B is required for HbF expression and F-cell formation, we used CRISPR-Cas9 targeting in HUDEP1 cells, which have high LIN28B levels and HbF expression, as well as in HUDEP2 cells, in which LIN28B expression is difficult to detect. We achieved up to 90% to 95% reduction in LIN28B protein in HUDEP1 cells and high editing efficiencies in HUDEP2 cells without a decrease in γ-globin protein (supplemental Figure 3A) or frequency of F cells. HUDEP2 cells tended to drift toward higher LIN28B and HbF expression when they had undergone a series of perturbations. When generating clonal LIN28B-deleted sublines, multiple nontargeted clones showed high LIN28B expression (supplemental Figure 3C) and high F-cell frequencies. However, multiple HUDEP2 clones with homozygous mutations predicted to ablate LIN28B expression still expressed HbF and had expected frequencies of F cells. Taken together, our data show that although increased LIN28B levels can drive higher HbF expression, the LIN28B/let-7 axis is not required for and likely does not account for F-cell formation in HUDEP2 cells.
In sum, stage-matched A- and F-HUDEP2 cells are nearly identical with regard to their proteome and transcriptome, suggesting that F cells do not likely represent a reversion to a globally fetal state, especially in light of the pronounced difference between native fetal and adult human erythroblasts. Moreover, key modulators of γ-globin transcription are expressed at similar levels in F and A erythroblasts and are either not relevant to the HUDEP2 F-cell phenotype or may be controlled posttranslationally.
F-cell analysis in primary human erythroid cultures
Healthy adults express small amounts of HbF in circulating F cells, and CD34+ hematopoietic stem and progenitor cell–derived erythroid culture systems also demonstrate a heterocellular distribution of HbF expression. To characterize F cells in primary samples, we performed a time-course RNA-seq experiment with cultures from 2 independent healthy donors using a 3-phase in vitro culture system. We chose time points at day 8, when we could first reliably sort F cells, and at days 11 and 14 for comparison with our previous work. We sorted matched F and A erythroblasts and validated the purification by qRT-PCR and western blotting for globins (Figure 3A). Analysis of globin transcript levels by ddPCR and protein levels by semiquantitative western blotting showed that day 11 F erythroblasts express a mixture of γ-globin and β-globin, similar to HUDEP2 cells and mature adult erythrocytes (supplemental Figure 4).11 Principal component analysis of RNA-seq data (Figure 3B) showed that samples clustered primarily by differentiation stage and by donor, but not by the presence of HbF. Using a significance threshold of 1.5-fold change and an adjusted value of P < .05, there were overall only a few differentially expressed transcripts at days 8 and 11 of differentiation (F-cell enriched: 25 transcripts at day 8, 8 transcripts at day 11, and 44 transcripts at day 14; A-cell enriched: 8 transcripts at day 8, 9 transcript at day 11, and 217 transcripts at day 14) (Figure 3C-D). There was also little overlap of the differentially expressed genes between different days of differentiation (Figure 3C), with only HBG1, HBG2, HBG1/2, and BGLT3 enriched across all 3 days. The number of differentially expressed transcripts was much smaller than differences we had reported between fetal and adult erythroblasts (at the same false discovery rate and fold-change threshold: 696 adult and 540 fetal transcripts at day 11 and 658 adult and 607 fetal transcripts at day 14).23 There was also little overlap of differentially expressed transcripts between F cells and fetal erythroblasts (3 transcripts at day 8, 3 transcripts at day 11, 29 transcripts at day 14). To ask in an unbiased fashion whether F cells represent a return to a fetal-like state, we carried out GSEA with gene sets of the top 250 fetal- or adult-enriched transcripts at days 11 or 14 of primary erythroid culture to probe our F-cell data sets from HUDEP2 and primary samples (supplemental Figure 5).23 In HUDEP2 samples, a fetal signature was enriched in F cells (normalized enrichment score [NES], 3.34) and an adult signature was negatively enriched (NES, −2.65), but few F-cell–enriched transcripts overlapped with fetal-specific transcripts. In primary cell cultures, a fetal signature was negatively enriched in day 8 F cells (NES, −1.65), whereas at days 11 and 14, only minimal enrichment of a fetal signature in F cells (NES, 1.59 at day 11 and 1.78 at day 14) was found but not at the magnitude seen in HUDEP2 cells. This analysis, together with low overlap between true fetal-specific and F-cell–enriched transcripts suggest that in primary erythroid cultures, F cells are not formed through reversion to a fetal state.
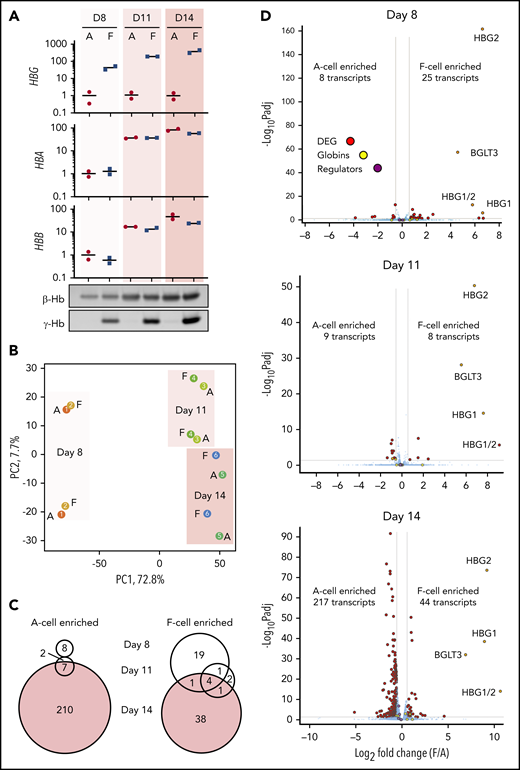
RNA-seq analysis of CD34+ HSPC-derived Fcells at different stages of differentiation. CD34+ cells from 2 independent donors were cultured, and stage-matched F cells were sorted on days 8, 11, and 14 of differentiation. (A) RT-PCR and western blot analysis of globins. RT-PCR data are normalized to mean expression of 3 housekeeping genes and shown relative to day 8 A cells as individual values and mean. (B) Principal component (PC) analysis of RNA-seq data from sorted F and A cells. Two biological replicates are shown for each time point. (C) Venn diagrams showing overlap of F-cell enriched or A-cell enriched transcripts between different days of differentiation. (D) RNA-seq data from days 8, 11, and 14 of differentiation. Average data from 2 replicates is shown. FDR <0.05; fold change >1.5. Globin transcripts and known HbF regulators (BCL11A, LRF, EIF2AK1, and SPOP) are highlighted.
RNA-seq analysis of CD34+ HSPC-derived Fcells at different stages of differentiation. CD34+ cells from 2 independent donors were cultured, and stage-matched F cells were sorted on days 8, 11, and 14 of differentiation. (A) RT-PCR and western blot analysis of globins. RT-PCR data are normalized to mean expression of 3 housekeeping genes and shown relative to day 8 A cells as individual values and mean. (B) Principal component (PC) analysis of RNA-seq data from sorted F and A cells. Two biological replicates are shown for each time point. (C) Venn diagrams showing overlap of F-cell enriched or A-cell enriched transcripts between different days of differentiation. (D) RNA-seq data from days 8, 11, and 14 of differentiation. Average data from 2 replicates is shown. FDR <0.05; fold change >1.5. Globin transcripts and known HbF regulators (BCL11A, LRF, EIF2AK1, and SPOP) are highlighted.
Although we matched cells on the basis of CD71 and CD235 markers as well as size, subtle differences in maturation might exist between F and A cells. To address this, we used GSEA with published transcriptome data sets across erythroid differentiation in CD34+ hematopoietic stem and progenitor cell–derived erythroid cultures.49 Cluster 1 and cluster 3 were derived from this data set by hierarchical clustering, and they consisted of transcripts that were continually up- and downregulated during differentiation, respectively. At day 8, there was little enrichment of either cluster (supplemental Figure 6), but at days 11 and 14, F-cell upregulated transcripts were positively enriched for cluster 3 and negatively enriched for cluster 1, suggesting a very mild maturation delay. Given the low overall magnitude of transcriptional changes and small magnitude of changes in selected erythroid maturation markers (supplemental Table 3), this delay is very subtle and unlikely to confound interpretation of the results.
Known HbF regulators are not differentially expressed in F erythroblasts
HbF expression in a subset of adult erythroid cells might be the result of changes in the levels of BCL11A and LRF, the predominant direct silencers of γ-globin transcription. Although a recent study suggested that developmental regulation of BCL11A levels occurs primarily at the level of translation,50 3 previous studies22-24 indicated that adult erythroblasts contain higher BCL11A mRNAs than their fetal counterparts, likely because of elevated transcription on the basis of RNA polymerase 2 chromatin immunoprecipitation sequencing experiments (supplemental Figure 7). Similar to our findings in HUDEP2 cells, primary erythroid cultures displayed no significant differences in BCL11A and LRF transcript levels as assessed by RNA-seq at day 8 of maturation (Figure 4A). None of the other known HbF regulators were significantly differentially expressed in F cells (supplemental Tables 4-6). We further examined BCL11A and LRF transcripts by qRT-PCR in sorted day 11 F and A cells from a total of 9 donors (Figure 4B), as well as by western blots from 3 donors (Figure 4C), and quantified mRNA expression of NuRD complex components in 9 donors (Figure 4D). There were no differences in expression of these molecules between F and A cells. Although we found increased LIN28B levels in HUDEP2 F cells, we were unable to detect LIN28B expression in F cells from primary cultured cells by RNA-seq, quantitative ddPCR, or single-molecule RNA fluorescence in situ hybridization (data not shown). We also targeted LIN28B using CRISPR-Cas9 with 2 guide RNAs in early stages of primary erythroid culture. Despite achieving high levels of editing with knockout mutations, we did not see a decrease in F cells under baseline conditions or with hydroxyurea or pomalidomide treatment (supplemental Figure 8). Hence, unexpectedly, changes in the levels of the known HbF regulators examined here do not account for elevated HbF production in primary F cells.
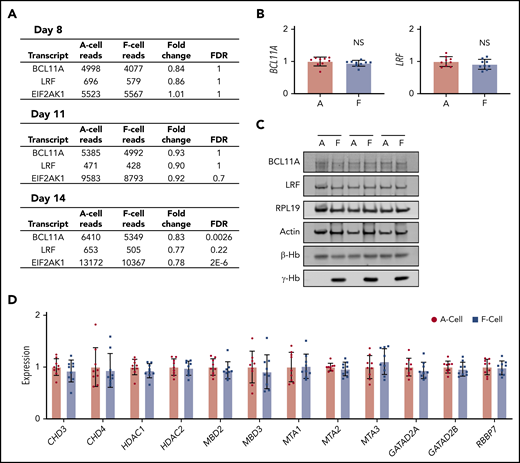
Known HbF regulators are not differentially expressed in CD34+ HSPC-derived Ferythroblasts. (A) RNA-seq transcript levels of selected known HbF regulators. Data are shown as average normalized reads from 2 biological replicates. (B) RT-PCR analysis of BCL11A and LRF expression from sorted day 11 F and A cells from a total of 9 donors across 3 experiments. Data are shown relative to A cells as mean ± SD. (C) Western blot analysis of HbF regulators from sorted day 11 F and A cells from 3 separate donors. (D) RT-PCR analysis of NuRD complex components from sorted day 11 F and A cells from a total of 9 donors across 3 experiments. Data are shown relative to A cells as mean ± SD.
Known HbF regulators are not differentially expressed in CD34+ HSPC-derived Ferythroblasts. (A) RNA-seq transcript levels of selected known HbF regulators. Data are shown as average normalized reads from 2 biological replicates. (B) RT-PCR analysis of BCL11A and LRF expression from sorted day 11 F and A cells from a total of 9 donors across 3 experiments. Data are shown relative to A cells as mean ± SD. (C) Western blot analysis of HbF regulators from sorted day 11 F and A cells from 3 separate donors. (D) RT-PCR analysis of NuRD complex components from sorted day 11 F and A cells from a total of 9 donors across 3 experiments. Data are shown relative to A cells as mean ± SD.
Pharmacologic HbF induction without reversion to a fetal-like state
Both hydroxyurea and the experimental HbF inducer pomalidomide augment HbF expression in a heterocellular fashion in vivo and ex vivo by poorly understood mechanisms.18,20,51-59 Previous studies profiled the transcriptional response to hydroxyurea treatment, but an open question remains regarding whether any changes in transcript levels are directly linked to HbF regulation. To test whether F cells generated in response to these compounds differ from those produced under baseline conditions, we purified matched F and A cells at day 11 after 5 days of culture in vehicle (∼20% F cells), hydroxyurea (∼30% F cells), and pomalidomide (∼40% F cells) from 2 healthy donors (Figure 5A). We also quantified globin transcripts by ddPCR and found that pomalidomide increased the fraction of γ-globin transcripts per F cell at the expense of β-globin transcripts (Figure 5B). We compared transcriptomes of F and A cells within each treatment group as well as between treatment groups (Figure 5C-D; supplemental Figure 9). Hydroxyurea and pomalidomide elicited significant transcriptional changes in both A and F cells when compared with corresponding cells from control samples (159 transcripts in A cells for hydroxyurea, 160 transcripts in F cells for hydroxyurea, 598 transcripts in A cells for pomalidomide, and 717 transcripts in F cells for pomalidomide); this is similar in magnitude to changes reported in patient reticulocytes after treatment with hydroxyurea (256 differentially expressed genes at a similar significance threshold).60 Remarkably, however, within each treatment condition, there were fewer transcriptional differences between F and A cells than there were between treatment conditions. Specifically, only 36 and 17 transcripts were enriched in hydroxyurea-treated A and F cells, respectively (supplemental Table 7). In the case of pomalidomide, more transcriptional changes were measured with 234 and 166 A- and F-cell–enriched mRNAs, respectively, but still markedly fewer changes than between treatment groups. GSEA using erythroid maturation data sets showed a mild differentiation delay in F cells in both hydroxyurea and pomalidomide treatments, but no greater enrichment of a fetal-like transcriptional signature than in untreated cultures (supplemental Figure 10). Overall, there was little overlap in transcripts differentially expressed across multiple conditions (Figure 5D) and no clear differences in F-cell expression of HbF regulators (supplemental Tables 8 and 9) or genes with variants associated with hydroxyurea response.61 In sum, the HbF response upon exposure to hydroxyurea and pomalidomide is not overtly associated with a reversion to a fetal-like state even though these compounds can trigger more widespread changes in gene expression.
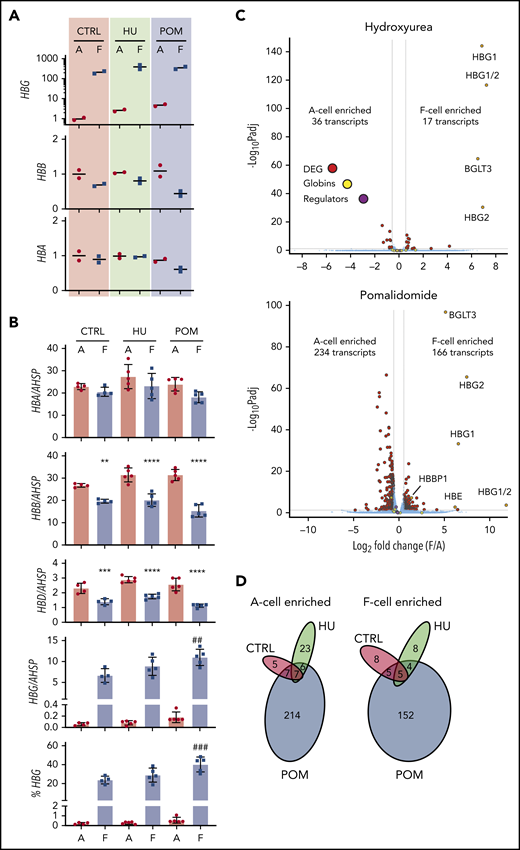
F-cell analysis after treatment with hydroxyurea and pomalidomide. Primary erythroid cultures from 2 donors were treated with vehicle (CTRL), hydroxyurea (HU), or pomalidomide (POM) starting on day 6 of culture. F and A erythroblasts were sorted on day 11 of culture. (A) RT-PCR analysis of globin transcript expression, normalized to mean expression of 3 housekeeping genes and shown relative to untreated A cells. (B) Globin transcript analysis by ddPCR in treated cells from a total of 5 donors. All graphs show individual data points as well as mean ± SD. **P < .01 vs A cells; ***P < .001 vs A cells; ****P < .0001 vs A cells; ##P < .01 vs control F cells; ###P < .001 vs control F cells by 1-way analysis of variance. (C) RNA-seq data comparing A and F cells in hydroxyurea (top) and pomalidomide (bottom) cultures. Average data from 2 replicates is shown. FDR <0.05; fold change >1.5. Globin transcripts and known HbF regulators (BCL11A, LRF, EIF2AK1, and SPOP) are highlighted. (D) Venn diagrams showing overlap of F-cell enriched or A-cell enriched transcripts between different treatment conditions.
F-cell analysis after treatment with hydroxyurea and pomalidomide. Primary erythroid cultures from 2 donors were treated with vehicle (CTRL), hydroxyurea (HU), or pomalidomide (POM) starting on day 6 of culture. F and A erythroblasts were sorted on day 11 of culture. (A) RT-PCR analysis of globin transcript expression, normalized to mean expression of 3 housekeeping genes and shown relative to untreated A cells. (B) Globin transcript analysis by ddPCR in treated cells from a total of 5 donors. All graphs show individual data points as well as mean ± SD. **P < .01 vs A cells; ***P < .001 vs A cells; ****P < .0001 vs A cells; ##P < .01 vs control F cells; ###P < .001 vs control F cells by 1-way analysis of variance. (C) RNA-seq data comparing A and F cells in hydroxyurea (top) and pomalidomide (bottom) cultures. Average data from 2 replicates is shown. FDR <0.05; fold change >1.5. Globin transcripts and known HbF regulators (BCL11A, LRF, EIF2AK1, and SPOP) are highlighted. (D) Venn diagrams showing overlap of F-cell enriched or A-cell enriched transcripts between different treatment conditions.
Discussion
In this study, we optimized protocols to directly compare HbF-expressing cells to matched HbF-low cells with an identical genetic background at multiple levels. Our data show that F erythroblasts are highly similar to non-HbF–expressing erythroblasts, with only a few transcriptional differences outside the β-globin locus. In this study, the magnitude of the differences between F and A erythroblasts was far less than that previously shown between fetal and adult erythroblasts. Moreover, the differentially expressed genes in F cells have minimal overlap with transcripts up- or downregulated in fetal cells. These findings demonstrate that F cells do not represent a reversion to a fetal-like state but rather a narrower set of changes confined to the β-globin locus. Of note, there was also no overlap between transcripts differentially expressed in F erythroblasts from HUDEP2 cells or primary cultures, suggesting either that different pathways account for HbF production between these cell systems or that none of the differentially expressed genes account for HbF expression.
An obvious model for F-cell formation would be that variable reduction in HbF co-regulator levels in a subset of cells accounts for de-repression of the γ-globin genes. Surprisingly, however, our data were inconsistent with this. One exception is LIN28B, which is higher in HUDEP2 F cells. LIN28B has a clearly defined role in the maintenance of a fetal state in multiple cell types42,43,45-47 and is capable of increasing HbF levels upon overexpression.44 However, our perturbative experiments suggest that LIN28B does not account for HbF increases in HUDEP2 F cells. In primary cell cultures, none of the known HbF regulators were significantly different in F vs A cells based on transcriptome studies. All previous studies but 1 found higher levels of BCL11A transcripts in adult erythroid cells compared with their counterparts, likely because of increased transcription.22-24,50 The lack of differences in BCL11A mRNA levels between F and A cells across multiple donor samples further supports the idea that F cells do not represent a reversal to a fetal-like state. Another possibility is that differences occur through posttranslational modifications or signaling events, which were not examined here, but we failed to find any clear transcriptional signatures that would point to a specific upstream signaling event.
Hydroxyurea has been in clinical use for more than 20 years, and yet the mechanism of its action on fetal hemoglobin is not understood; some proposed pathways include modification of nitric oxide–mediated signaling and cell cycle–related changes.54-56 Pomalidomide is a more recently developed anti-neoplastic medication with demonstrated fetal hemoglobin induction potentially through multiple regulators.57-59 Previous studies have attempted to clarify the mechanisms of these drugs by comparing gene and protein expression between treated and untreated cells. With regard to HbF regulation, we believe a more informative comparison is that between drug-exposed cells with high and low HbF levels. Thus, the transcriptional differences between F and A erythroblasts were smaller than those resulting from the drug treatment itself when compared with controls. We found few differentially expressed transcripts that were common to both drugs, suggesting that they work through different pathways. Another interesting finding is that HbF inducers can differ in whether they increase the number of F cells, the amount of HbF per F cell, or both. Pomalidomide is one of the most potent experimental HbF inducers, and it nearly doubled both the cellular γ-globin fraction and the number of F cells. At the transcript level, there were no clear changes resulting from either hydroxyurea or pomalidomide that would account for HbF induction, and further studies of epigenetic and signaling pathways will be needed to assess the mechanism of HbF induction by these compounds.
It is clear that F cells express a mixture of γ-globin and β-globin rather than revert to a state of pure fetal globin expression. β-globin and γ-globin transcription are controlled through developmental stage–specific changes in contacts with the locus control region, and these interactions are dynamic.62,63 The results in this study suggest that the primary differences between F and A cells occur at the β-globin locus. Future imaging and chromatin configuration studies will characterize F cells at the epigenetic and transcriptional bursting level under different conditions; our methods will allow for further interrogation of F cells generated in response to different stimuli and will further help researchers understand regulation of globin switching.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Florin Tuluc and the staff of the Children’s Hospital of Philadelphia Flow Cytometry Core for assistance with cell sorting optimization, R. Kurita and Y. Nakamura for contributing the HUDEP1 and HUDEP2 cells, and the DiGaetano family for their generous support.
This work was supported by National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases grants DK054937 (G.A.B.) and DK106766 (G.A.B. and R.C.H.), NIH National Heart, Lung, and Blood Institute grant HL119479 (G.A.B.) and T32 training grant HL007150-42 (E.K.), the American Society of Hematology Research Training Award for Fellows (E.K.), and the St. Jude Children’s Research Hospital Collaborative Research Consortium on Novel Gene Therapies for Sickle Cell Disease.
Authorship
Contribution: E.K. and G.A.B. conceived the project, designed the experiments, and wrote the manuscript; E.K., P.H., S.A.P., O.A., B.M.G., and C.A.K. carried out experiments and analyzed data; M.S. carried out experiments; E.K., Z.Z., R.C.H., and G.A.B. prepared the manuscript; and all authors were involved in the critical revision of the manuscript, had full access to the data in this study, reviewed drafts of the manuscript, and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Eugene Khandros, The Children’s Hospital of Philadelphia, Division of Hematology, Abramson Research Center 316A, 3615 Civic Center Blvd, Philadelphia, PA 19104; e-mail: khandrose@email.chop.edu.
