

Key Points
MDS/AML are the most common short telomere malignancies; their natural history is complicated by extra-hematopoietic disease.
Adults with short telomeres who have no MDS/AML have premature age-related clonal hematopoiesis.

Abstract
Short telomeres have been linked to cancer risk, yet other evidence supports them being tumor suppressive. Here, we report cancer outcomes in individuals with germline mutations in telomerase and other telomere-maintenance genes. Among 180 individuals evaluated in a hospital-based setting, 12.8% had cancer. Solid tumors were rare (2.8%); nearly all were young male DKC1 mutation carriers, and they were generally resectable with good short-term outcomes. Myelodysplastic syndrome (MDS) was most common, followed by acute myeloid leukemia (AML); they accounted for 75% of cancers. Age over 50 years was the biggest risk factor, and MDS/AML usually manifested with marrow hypoplasia and monosomy 7, but the somatic mutation landscape was indistinct from unselected patients. One- and 2-year survival were 61% and 39%, respectively, and two-thirds of MDS/AML patients died of pulmonary fibrosis and/or hepatopulmonary syndrome. In one-half of the cases, MDS/AML patients showed a recurrent peripheral blood pattern of acquired, granulocyte-specific telomere shortening. This attrition was absent in age-matched mutation carriers who did not have MDS/AML. We tested whether adult short telomere patients without MDS/AML also had evidence of clonal hematopoiesis of indeterminate potential–related mutations and found that 30% were affected. These patients also primarily suffered morbidity from pulmonary fibrosis during follow-up. Our data show that the Mendelian short telomere syndromes are associated with a relatively narrow cancer spectrum, primarily MDS and AML. They suggest that short telomere length is sufficient to drive premature age-related clonal hematopoiesis in these inherited disorders.
Introduction
Short telomere length is genetically determined; it is also acquired with aging.1 In some animal models, short telomeres have been associated with increased cancer risk, but other evidence supports them being tumor suppressive.1 Initial studies of telomerase-null mice with short telomeres, bred on a mixed strain background, suggested that telomere shortening drives genome instability via chromosome end-to-end fusions.2 Short telomere mice that lacked Tp53 also had increased tumor formation.3 However, more recently, outbred telomerase-null mice with humanized telomere length showed no evidence of genome instability or overt cancer predisposition.4,5 Moreover, because most humans with short telomeres are TP53 competent, the relevance of the earlier animal models to understanding the role of short telomere length in human cancer risk has been unclear. Adding to the conflicting evidence, tumor-prone short telomere mice that are Tp53 competent have significantly improved survival rates compared with their long telomere counterparts.1,6-9 Epidemiologic studies have also differed in their conclusions on the role of short telomeres in cancer risk, with some pointing to a tumor-promoting role10 ; other recent literature more emphatically indicates the opposite: that long telomeres are associated with increased risk of common cancers.1,11
Germline mutations in telomerase and other telomere-maintenance genes manifest in humans in a spectrum of short telomere syndromes that span the entire age range.12 They are the most common of the premature aging syndromes.1 Infants and young adults present with degenerative phenotypes in high turnover tissues including immunodeficiency, aplastic anemia, and gastrointestinal disease.13-15 Children may also show, in more rare cases, features of dyskeratosis congenita, a disorder classically defined by a triad of mucocutaneous findings: oral leukoplakia, nail dystrophy, and skin hyperpigmentation.16,17 The majority of patients carrying mutant telomerase and telomere-maintenance genes present in adulthood with isolated pulmonary fibrosis with or without emphysema.18,19 Studies of cancer in short telomere patients have been limited to pediatric dyskeratosis congenita patients,16,20,21 and the overall prevalence, risk factors, and clinical course are yet to be defined. Here, we report on a 16-year experience with cancer, including its treatment outcomes, in an adult-predominant short telomere syndrome population that was followed in a hospital-based setting.
Methods
Subjects, study design, and consent
From 1 July 2003 to 30 June 2019, as part of the Telomere Syndrome Registry, we recruited subjects from patients evaluated at Johns Hopkins Hospital or referring centers.14,22 They were assigned the short telomere syndrome diagnosis if they: (1) carried a validated pathogenic mutation in telomerase or another telomere-maintenance gene, (2) had short telomere length with classic features of a familial short telomere syndrome as defined,15 or (3) had classic short telomere syndrome features with abnormally low telomerase RNA levels, in the presence or absence of a family history, as previously defined.15,23,24 Dyskeratosis congenita was considered the diagnosis in patients who had at least 1 of the 3 classic mucocutaneous triad features. All consecutive individuals fulfilling the prespecified inclusion criteria were included in this analysis. The study was approved by the Johns Hopkins Medicine Institutional Review Board, and all subjects gave written informed consent. Follow-up was last assessed on 1 September 2019.
Germline sequencing and mutation analysis
Genomic DNA was isolated from blood using the EZ1 DNA Blood 350 µL kit (Qiagen). Mutations were identified by exome or genome sequencing,23,25 panel-based testing,18 or from clinical sequencing studies. Mutations were deemed pathogenic based on family segregation and/or functional studies as previously described.23,24,26 For the first 100 recruited patients, mutations were previously published.27 Previously unreported mutations are listed in supplemental Table 1 (available on the Blood Web site), and all new mutations are deposited into the Telomerase Database (telomerase.asu.edu).28
Telomere length measurement
Telomere length was measured on peripheral blood by flow cytometry and fluorescence in situ hybridization (flowFISH), as previously described.27,29 The Δ telomere length, for lymphocytes and granulocytes, was calculated as the difference of the telomere length from the respective age-adjusted median as described previously.30
Clinical review of cancer data
We curated noncutaneous cancer diagnoses from the clinical history, all verified with primary pathology reports. We used the most recent tumor, node, metastasis (TNM) staging to classify the solid tumors,31 and the 2016 revised World Health Organization (WHO) classification for myeloid neoplasms.32 We reviewed primary medical records for the treatment course and outcomes. Bone marrow pathology for myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML), where available, was reviewed centrally by a hematopathologist (A.S.D.). Next-generation sequencing and analyses were also performed centrally in a clinical laboratory (Johns Hopkins Genomics). Clinical data were reviewed and adjudicated by K.E.S. and M.A. Cancers that were deemed transplant-related were excluded: 1 donor-derived lung adenocarcinoma, 2 metastatic squamous cell cancers, 1 metastatic small cell cancer (all after lung transplantation), and an early stage head and neck cancer diagnosed 5 years after hematopoietic stem cell transplantation (HSCT) in a dyskeratosis congenita patient.
SEER comparisons
We compared the number of MDS, AML, and oral cancers (anterior tongue/gingival) observed in our cohort of 180 individuals with the number of expected diagnoses over the same follow-up time using the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program data (www.seer.cancer.gov) as previously described.33 Briefly, we used SeerStat version 8.3.6 statistical software to calculate age-adjusted incidence rates of AML and oral cancers separately for men and women, according to race (white, black, other), and by year from 1975 through 2016; rates for the 2017 to 2019 period were extrapolated using a regression model. Rates for MDS were also calculated for the same subgroups by year from 2001 to 2016 and extrapolated for 2017 to 2019. For each individual, we calculated the cumulative sum of the yearly incidence rate beginning with either: (1) the individual’s birth year or (2) 1975 for oral cancers and AML, and 2001 for MDS if the individual was born before 2001, and up through the year of last known follow-up or year of diagnosis, if affected. For each cancer, we took the sum of these values across all individuals as the expected number of diagnoses for this cohort. The observed-to-expected (O/E) ratio was calculated as the sum of the observed diagnoses in the Johns Hopkins cohort divided by the expected number in SEER. The 95% confidence interval (CI) was calculated using a bootstrap approach with 10 000 simulations. As a sensitivity analysis, because the follow-up time for MDS is limited to 2001 and later, the yearly incidence rates for MDS in 2001 were backfilled through to 1975 and the expected number of MDS cases was recalculated.
Targeted sequencing for MDS/AML somatic mutations
Library preparation
We performed somatic mutation analysis on peripheral blood using a custom-designed targeted panel as described.34 Briefly, DNA hybrid capture libraries were prepared using an Agilent SureSelect-XT target enrichment kit with 200 ng of peripheral blood–derived genomic DNA template as input. The somatic MDS/AML genes queried are listed in supplemental Table 2.
Somatic sequencing
Sequencing was performed using a HiSeq 2500 (Illumina) platform using paired-end 200-bp Rapid Run v2 chemistry. Reads were aligned to the GRCh37 reference genome using the Burrows-Wheeler aligner (v.0.7.10).35 Variants were called and annotated directly from the BAM file using a custom variant caller pipeline (MDLVC v.6) that had multiple filtering steps as described in Zheng et al.34 Mean depth of coverage was 747× (range, 376-1296) and mean on-target coverage was 98.9% at >150×. Variants with ≥2% variant allele frequency (VAF) were confirmed by manual inspection by K.E.S. and L.H. Variants with VAF 40% to 60% were considered germline and excluded.
Somatic variant interpretation
Nonsynonymous variants with VAF <40% or >60% were further analyzed and cataloged if they: (1) resulted in a frameshift or nonsense, (2) were present in COSMIC (v82), or (3) were novel (ie, absent from both COSMIC and dbSNP [v150]). To ensure that variants studied were not common polymorphisms, we also excluded them if they had a frequency of >0.05% in gnomAD (r2.0.2, last accessed 2 January 2019).
Statistical analysis
Statistical analyses that are not SEER-related and their respective graphics were prepared using GraphPad Prism 6.0 (GraphPad Software).
Results
MDS/AML are the most common cancers in the short telomere syndromes
We assessed cancer diagnoses among 180 consecutively recruited individuals from 113 families who enrolled in the Johns Hopkins Telomere Syndrome Registry from 2003 to 2019 (Figure 1A; Table 1). Subjects represented a wide age range, from 1 to 81 years, with a median age of 50 years; 83% were adults and 59% of subjects were male (Table 1). Seventy-nine percent carried a pathogenic mutation in 1 of 8 telomere-related genes, and the remaining individuals had classic short telomere syndrome features with a positive family history and/or telomerase RNA insufficiency (Figure 1A; Table 1). Germline mutations in TERT were most common (42%) followed by TR, RTEL1, DKC1, PARN, TIN2, NAF1, and ZCCHC8 (Figure 1A; Table 1). The vast majority, 92%, had no features of dyskeratosis congenita (Table 1). Within this cohort, we documented 24 noncutaneous cancers in 23 patients, reflecting a 12.8% incidence (Figure 1B; Tables 2 and 3). MDS and AML were most common and accounted for 75% of short telomere cancers (Figure 1C). The observed number of 14 MDS cases was 145-fold higher than would be expected in a similar cohort with the same distribution of age, race, and sex (95% CI, 81-216; P < .001; Mann-Whitney U test). The O/E ratio for AML was 21 (95% CI, 4-31; P = .025). Age was the biggest risk factor with most MDS/AML diagnosed after 50 years (4 of 90 <50 years vs 14 of 90 ≥50 years; odds ratio [OR], 4.0; 95% CI, 1.2-12.5). The median age of 53 years at MDS/AML diagnosis is 2 decades younger than the US population.36,37 Solid tumors were overall rare, diagnosed in 2.8%. Six cancers were identified in 5 patients and oral cavity squamous cancers represented one-half of these cases. The O/E ratio for oral cavity cancers was 61 (95% CI, 0-140; P = .08). The age at diagnosis of solid tumors was younger than MDS/AML (median, 35 vs 53 years; P = .01; Mann-Whitney U test; Figure 1D), and most were diagnosed prior to 40 years of age (Figure 1D). These data indicated that although the overall rate of cancer is relatively low in short telomere patients, it is significantly higher than population rates and the risk is highest for MDS.
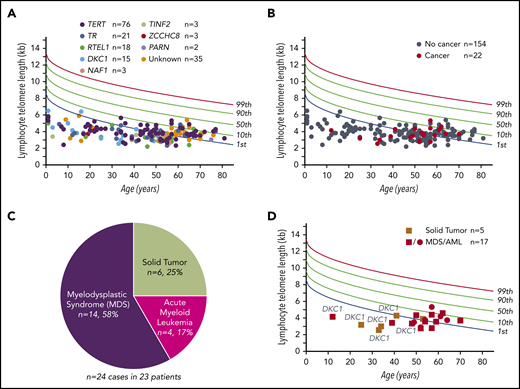
Prevalence and cancer phenotypes in patients with germline defects in telomere maintenance. (A) Telogram of lymphocyte telomere lengths of 176 short telomere syndrome patients relative to a nomogram of healthy controls with percentile lines labeled on the right. The mutant gene for each subject is color coded and the key is shown above the chart. Four patients included in this study were deceased prior to telomere-length measurement. (B) Telogram showing the patients with noncutaneous cancer diagnoses within the Johns Hopkins cohort of 176 patients in this study denoted in red; individuals without cancer are denoted in gray. One MDS/AML patient (among 23 with cancer) is not shown because of missing telomere-length data as explained in panel A. (C) Distribution of the 24 cancer types identified in 23 patients. (D) Telogram showing solid tumor and MDS/AML cancers by age and sex. The squares refer to male subjects and circles to female subects. DKC1 mutation carriers are annotated to show their young-onset disease and predilection to solid tumors.
Prevalence and cancer phenotypes in patients with germline defects in telomere maintenance. (A) Telogram of lymphocyte telomere lengths of 176 short telomere syndrome patients relative to a nomogram of healthy controls with percentile lines labeled on the right. The mutant gene for each subject is color coded and the key is shown above the chart. Four patients included in this study were deceased prior to telomere-length measurement. (B) Telogram showing the patients with noncutaneous cancer diagnoses within the Johns Hopkins cohort of 176 patients in this study denoted in red; individuals without cancer are denoted in gray. One MDS/AML patient (among 23 with cancer) is not shown because of missing telomere-length data as explained in panel A. (C) Distribution of the 24 cancer types identified in 23 patients. (D) Telogram showing solid tumor and MDS/AML cancers by age and sex. The squares refer to male subjects and circles to female subects. DKC1 mutation carriers are annotated to show their young-onset disease and predilection to solid tumors.
Characteristics of Johns Hopkins Telomere Syndrome Registry subjects
| n = 180 | |
|---|---|
| No. of unrelated families | 113 |
| Male, no. (%) | 106 (59) |
| Female, no. (%) | 74 (41) |
| Classic dyskeratosis congenita,* no. (%) | 14 (7.8) |
| Telomere length by flowFISH, no. (%) | 176 (98) |
| Age range, y, no. (%) | |
| 0-20 | 30 (17) |
| 21-40 | 31 (17) |
| 41-60 | 78 (43) |
| 61-80 | 40 (22) |
| >80 | 1 (1) |
| Mutant gene,†no. (%) | |
| TERT | 76 (42.2) |
| TR | 22 (12.2) |
| RTEL1 | 18 (10.0) |
| DKC1 | 15 (8.3) |
| NAF1 | 3 (1.7) |
| TINF2 | 3 (1.7) |
| PARN | 2 (1.1) |
| ZCCHC8 | 3 (1.7) |
| Unknown | 38 (21.1) |
| n = 180 | |
|---|---|
| No. of unrelated families | 113 |
| Male, no. (%) | 106 (59) |
| Female, no. (%) | 74 (41) |
| Classic dyskeratosis congenita,* no. (%) | 14 (7.8) |
| Telomere length by flowFISH, no. (%) | 176 (98) |
| Age range, y, no. (%) | |
| 0-20 | 30 (17) |
| 21-40 | 31 (17) |
| 41-60 | 78 (43) |
| 61-80 | 40 (22) |
| >80 | 1 (1) |
| Mutant gene,†no. (%) | |
| TERT | 76 (42.2) |
| TR | 22 (12.2) |
| RTEL1 | 18 (10.0) |
| DKC1 | 15 (8.3) |
| NAF1 | 3 (1.7) |
| TINF2 | 3 (1.7) |
| PARN | 2 (1.1) |
| ZCCHC8 | 3 (1.7) |
| Unknown | 38 (21.1) |
Classic dyskeratosis congenita was defined as having at least 1 of the mucocutaneous triad features.
Three patients had biallelic mutations: RTEL1 (n = 2) and TERT (n = 1); these were counted each as 1 mutation.
Characteristics and treatment course of short telomere solid tumor patients (n = 6 cases)
| ID | Age, y | M/F | Gene | Mutation | Cancer | Stage | Therapy | Follow-up, mo |
|---|---|---|---|---|---|---|---|---|
| 1* | 25 | M | DKC1 | Pro409Arg | SCC tongue | Stage I | Resection | 20 |
| 2* | 35 | M | DKC1 | Leu317Phe | SCC tongue | Stage II | Resection | 96 |
| 3* | 33 | M | DKC1 | IVS2-5C>G | SCC anal | Stage I | Resection, 5FU topical, Pembro, radiation | 4† |
| 36 | SCC oral cavity | Stage I | Resection | 16 | ||||
| 4* | 38 | M | DKC1 | Ala353Val | Adenocarcinoma rectal | Stage IIIA | Resection, FOLFOX | 24 |
| 5 | 53 | M | TERT | Ile686Met | SCC anal | Stage IIIB | Radiation | 30 |
| ID | Age, y | M/F | Gene | Mutation | Cancer | Stage | Therapy | Follow-up, mo |
|---|---|---|---|---|---|---|---|---|
| 1* | 25 | M | DKC1 | Pro409Arg | SCC tongue | Stage I | Resection | 20 |
| 2* | 35 | M | DKC1 | Leu317Phe | SCC tongue | Stage II | Resection | 96 |
| 3* | 33 | M | DKC1 | IVS2-5C>G | SCC anal | Stage I | Resection, 5FU topical, Pembro, radiation | 4† |
| 36 | SCC oral cavity | Stage I | Resection | 16 | ||||
| 4* | 38 | M | DKC1 | Ala353Val | Adenocarcinoma rectal | Stage IIIA | Resection, FOLFOX | 24 |
| 5 | 53 | M | TERT | Ile686Met | SCC anal | Stage IIIB | Radiation | 30 |
5FU, 5-fluoruracil; F, female; FOLFOX, 5FU, oxaliplatin leukovorin regimen; M, male; Pembro, pembrolizumab; SCC, squamous cell carcinoma.
These 4 patients had classic mucocutaneous features of dyskeratosis congenita.
This patient had multiple local recurrences first detected 4 months after initial resection requiring multiple lines of therapy as listed. He had human papilloma virus–positive disease and a concurrent severe telomere-related T-cell immunodeficiency.
Clinical characteristics of short telomere syndrome–mediated MDS/AML
| Characteristic | (n = 18) |
|---|---|
| Median age at diagnosis, y | 53 (12-71) |
| Male sex, no. (%) | 12 (67) |
| Mutant gene | |
| TERT | 3 |
| RTEL1 | 3 |
| DKC1 | 2 |
| TR | 1 |
| NAF1 | 1 |
| Unknown | 8 |
| Myeloid neoplasm, no. (%) | |
| MDS | 14 (78) |
| AML arising from MDS | 3 (17) |
| Treatment-related AML | 1 (6) |
| Bone marrow cellularity for MDS,* n = 14,no. (%) | |
| Hypocellular | 7 (50) |
| Patchy (both hyper/hypocellular) | 0 (0) |
| Normocellular | 3 (21) |
| Hypercellular | 4 (29) |
| Bone marrow cellularity for AML, n = 4, no. (%) | |
| Hypocellular | 1 (25) |
| Patchy (both hyper/hypocellular) | 2 (50) |
| Normocellular | 1 (25) |
| Hypercellular | 0 (0) |
| Cytogenetics, MDS/AML, no. (%) | |
| Monosomy 7 alone | 3 (17) |
| Monosomy 7 with another abnormality | 7 (39) |
| Other abnormality | 7 (38.5) |
| Unknown | 1 (5.5) |
| Characteristic | (n = 18) |
|---|---|
| Median age at diagnosis, y | 53 (12-71) |
| Male sex, no. (%) | 12 (67) |
| Mutant gene | |
| TERT | 3 |
| RTEL1 | 3 |
| DKC1 | 2 |
| TR | 1 |
| NAF1 | 1 |
| Unknown | 8 |
| Myeloid neoplasm, no. (%) | |
| MDS | 14 (78) |
| AML arising from MDS | 3 (17) |
| Treatment-related AML | 1 (6) |
| Bone marrow cellularity for MDS,* n = 14,no. (%) | |
| Hypocellular | 7 (50) |
| Patchy (both hyper/hypocellular) | 0 (0) |
| Normocellular | 3 (21) |
| Hypercellular | 4 (29) |
| Bone marrow cellularity for AML, n = 4, no. (%) | |
| Hypocellular | 1 (25) |
| Patchy (both hyper/hypocellular) | 2 (50) |
| Normocellular | 1 (25) |
| Hypercellular | 0 (0) |
| Cytogenetics, MDS/AML, no. (%) | |
| Monosomy 7 alone | 3 (17) |
| Monosomy 7 with another abnormality | 7 (39) |
| Other abnormality | 7 (38.5) |
| Unknown | 1 (5.5) |
All of the normocelluar and hypercellular MDS patients (7 of 7) had ring sideroblasts in >15% of cells examined.
The risk of solid cancers is higher in male DKC1 mutation carriers
We examined whether there were correlations between certain mutant genes and cancer risk. We found no signal for MDS/AML (supplemental Table 3). However, nearly all of the solid tumor patients were male and DKC1 mutation carriers who had classic features of dyskeratosis congenita (80%; 4 of 5) (Figure 1A; Tables 2 and 3). The risk of solid tumors was higher in DKC1 mutation carriers (4 of 15 vs 1 of 165 non-DKC1 mutation carriers; OR, 60; 95% CI, 8.0-729.0). This genotype-phenotype correlation was telomere length independent as there was no difference between DKC1 and non-DKC1 subjects (P = .5). DKC1 mutation carriers were also generally more cancer-prone beyond solid tumors with an overall cancer rate of 40% vs 11% in non-DKC1 mutation carriers (6 of 15 vs 17 of 165; OR, 5.8; 95% CI, 1.7-19.0).
In general, short-term cancer outcomes with solid tumors were favorable. Nearly all of the solid cancers, which were generally mucosal and of squamous histology, were resectable at diagnosis and all 5 solid tumor patients are alive at follow-up (median, 24 months; range, 20-96 months; Table 2). Only 1 patient recurred; he had a refractory course of locally advanced anal squamous cell cancer associated with condylomatous human papilloma virus–associated carcinoma in situ and in the background of a severe telomere-related T-cell immunodeficiency13 (Table 2).
Distinctive clinicopathologic features of telomere-mediated MDS/AML
We next tested whether telomere-mediated MDS/AML had distinguishing clinical or pathologic features. Most patients reported a family history of pulmonary fibrosis, liver disease, or aplastic anemia at diagnosis (61%; 11 of 18). In only a small subset was there a first-degree relative who had a history of MDS/AML (17%; 3 of 18). One patient had no personal or family history of short telomere syndrome features; he carried a de novo DKC1 mutation. The most common comorbidity was pulmonary fibrosis, which preceded MDS/AML diagnosis in two-thirds of cases (67%; 12 of 18). The median time from pulmonary fibrosis to MDS/AML diagnosis was 3.0 years (range, 2 months to 15 years). These data indicated that although some patients with telomere-mediated MDS/AML may present with isolated disease, a majority have short telomere comorbidities at diagnosis, especially pulmonary fibrosis.
We next examined the pathological features of MDS, but found that no single distinct WHO subtype predominated. Classifications ranged from MDS-unclassified (cytogenetic abnormality but no overt dysplasia; n = 2), MDS with single-lineage dysplasia (n = 1), MDS with multilineage dysplasia (n = 2), MDS with ring sideroblasts (n = 4), to MDS with excess blasts (n = 5) (Table 3). Marrow pathology in these MDS patients, however, revealed several features that are otherwise rare in sporadic disease (Table 3). In half (50%; 7 of 14), the marrow was hypocellular (Figure 2A-C); this rate is higher than the 10% to 15% quoted for the general MDS population.38 For the other half with normocellular or hypercellular MDS, ring sideroblasts were documented in all cases (7 of 7, defined as >15% of erythroid precursors, 4 with MDS with ring sideroblasts and 3 with excess blasts) (Figure 2D-H). This prevalence is also higher than the 10% to 15% rate reported for sporadic MDS.39 As outlined further in the following sections, only 2 of the 7 ring sideroblast–positive patients had mutations in SF3B1 (Figure 2R). Three of the 4 AML patients also had background dysplasia and all 3 showed distinctively hypocellar or patchy-cellular marrows (Figure 2I-P; Table 3). Regardless of marrow cellularity, all of the MDS/AML patients had hypoproliferative disease with cytopenias at diagnosis and throughout their course. The high rate of hypoplasia and hypoproliferation suggested that these neoplasms arise on a continuum with aplastic anemia.
![Histopathologic and molecular characterization of short telomere MDS/AML. (A) Hypoplastic MDS (MDS with multilineage dysplasia; biopsy [hematoxylin-and-eosin stain (H&E); original magnification ×10]). (B) Multilineage dysplasia involving the myeloid (red arrows), erythroid (green arrow; aspirate [Wright-Giemsa stain; original magnification ×100]) and (C) megakaryocytic (yellow arrows; biopsy [H&E; original magnification ×40]) lineages. (D) MDS (MDS with ring sideroblasts and multilineage dysplasia; biopsy [H&E; original magnification ×10]). (E) Multilineage dysplasia involving the erythroid lineage (green arrows; aspirate [Wright-Giemsa stain; original magnification ×100]) with (F) abundant ring sideroblasts (aspirate [iron stain; original magnification ×100x]). Rare blasts (G, black arrow; Wright-Giemsa stain; original magnification ×100) are seen, and megakaryocytes (H) are also dysplastic (yellow arrows; H&E; original magnification ×40). (I) Hypoplastic AML (AML with myelodysplasia-related changes; biopsy [H&E; original magnification ×10]). Increased blasts (J, ∼70%) with background multilineage dysplasia involving the myeloid (K, red arrows) and erythroid (L, green arrows) lineages (aspirate [Wright-Giemsa stain; original magnification ×100]). (M) Hypoplastic AML (AML with myelodysplasia-related changes; biopsy [H&E; original magnification ×10]). Increased blasts (N; ∼50%) with background multilineage dysplasia involving the myeloid (O, red arrows) and erythroid (P, green arrow) lineages (aspirate [Wright-Giemsa stain; original magnification ×100]). (Q) Pie chart showing the distribution of karyotypes of 18 MDS/AML cases. Monosomy 7 abnormalities are listed on the left. (R) Somatic mutation profile of age-matched short telomere patients separated by those with (9 male/5 female) and without (11 male/9 female) MDS/AML. The age for each patient is included on top as is the presence of a monosomy 7 abnormality [−7, del(7q) or der(1;7)(q10;p10)]. All sequencing was performed on blood, except for the 2 AML cases (denoted as bolded ages) for which bone marrow was used. The ring sideroblasts (defined as >15% of cells examined) are denoted below the MDS/AML cases by an asterisk (*). Of note, another patient (of a total of 7) had ring sideroblasts but was not included in this analysis because of insufficient DNA. The respective germline mutations for each case are annotated in supplemental Figure 1A.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/22/10.1182_blood.2019003264/5/m_bloodbld2019003264f2.png?Expires=1767710937&Signature=IcjqKDjf-T7Zz1XONTAg6CuO9FceIVb1K3xFxQJI8GTP4WLWWWSMCq3M-TLtomVptzpqvCyp2GdOQFA5JaX5aF5HqqbR798ezVC6Z3ObdKfDIitBYPWdJ2-y7OP0vCTpnVjJ6vagzEOO9hU~zMMPQ8S6UdCVIRAEy3tO49YdPoN5vDmJfuj7nweZRFUDhhr8KTer1l3rXYO2Th-t2fgtloJAEH4kVxTduZz4jOLi3MoQVGUTaA9qJi0ihK8S1Vy7UbSdcRQJmpgl12tTefA0rqG1evloHTqbqtwjk1gqzWDs6MF0mV3euLmRKHFijgsNx8GpCC4d-QClh9SSO5qxpg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Histopathologic and molecular characterization of short telomere MDS/AML. (A) Hypoplastic MDS (MDS with multilineage dysplasia; biopsy [hematoxylin-and-eosin stain (H&E); original magnification ×10]). (B) Multilineage dysplasia involving the myeloid (red arrows), erythroid (green arrow; aspirate [Wright-Giemsa stain; original magnification ×100]) and (C) megakaryocytic (yellow arrows; biopsy [H&E; original magnification ×40]) lineages. (D) MDS (MDS with ring sideroblasts and multilineage dysplasia; biopsy [H&E; original magnification ×10]). (E) Multilineage dysplasia involving the erythroid lineage (green arrows; aspirate [Wright-Giemsa stain; original magnification ×100]) with (F) abundant ring sideroblasts (aspirate [iron stain; original magnification ×100x]). Rare blasts (G, black arrow; Wright-Giemsa stain; original magnification ×100) are seen, and megakaryocytes (H) are also dysplastic (yellow arrows; H&E; original magnification ×40). (I) Hypoplastic AML (AML with myelodysplasia-related changes; biopsy [H&E; original magnification ×10]). Increased blasts (J, ∼70%) with background multilineage dysplasia involving the myeloid (K, red arrows) and erythroid (L, green arrows) lineages (aspirate [Wright-Giemsa stain; original magnification ×100]). (M) Hypoplastic AML (AML with myelodysplasia-related changes; biopsy [H&E; original magnification ×10]). Increased blasts (N; ∼50%) with background multilineage dysplasia involving the myeloid (O, red arrows) and erythroid (P, green arrow) lineages (aspirate [Wright-Giemsa stain; original magnification ×100]). (Q) Pie chart showing the distribution of karyotypes of 18 MDS/AML cases. Monosomy 7 abnormalities are listed on the left. (R) Somatic mutation profile of age-matched short telomere patients separated by those with (9 male/5 female) and without (11 male/9 female) MDS/AML. The age for each patient is included on top as is the presence of a monosomy 7 abnormality [−7, del(7q) or der(1;7)(q10;p10)]. All sequencing was performed on blood, except for the 2 AML cases (denoted as bolded ages) for which bone marrow was used. The ring sideroblasts (defined as >15% of cells examined) are denoted below the MDS/AML cases by an asterisk (*). Of note, another patient (of a total of 7) had ring sideroblasts but was not included in this analysis because of insufficient DNA. The respective germline mutations for each case are annotated in supplemental Figure 1A.
Histopathologic and molecular characterization of short telomere MDS/AML. (A) Hypoplastic MDS (MDS with multilineage dysplasia; biopsy [hematoxylin-and-eosin stain (H&E); original magnification ×10]). (B) Multilineage dysplasia involving the myeloid (red arrows), erythroid (green arrow; aspirate [Wright-Giemsa stain; original magnification ×100]) and (C) megakaryocytic (yellow arrows; biopsy [H&E; original magnification ×40]) lineages. (D) MDS (MDS with ring sideroblasts and multilineage dysplasia; biopsy [H&E; original magnification ×10]). (E) Multilineage dysplasia involving the erythroid lineage (green arrows; aspirate [Wright-Giemsa stain; original magnification ×100]) with (F) abundant ring sideroblasts (aspirate [iron stain; original magnification ×100x]). Rare blasts (G, black arrow; Wright-Giemsa stain; original magnification ×100) are seen, and megakaryocytes (H) are also dysplastic (yellow arrows; H&E; original magnification ×40). (I) Hypoplastic AML (AML with myelodysplasia-related changes; biopsy [H&E; original magnification ×10]). Increased blasts (J, ∼70%) with background multilineage dysplasia involving the myeloid (K, red arrows) and erythroid (L, green arrows) lineages (aspirate [Wright-Giemsa stain; original magnification ×100]). (M) Hypoplastic AML (AML with myelodysplasia-related changes; biopsy [H&E; original magnification ×10]). Increased blasts (N; ∼50%) with background multilineage dysplasia involving the myeloid (O, red arrows) and erythroid (P, green arrow) lineages (aspirate [Wright-Giemsa stain; original magnification ×100]). (Q) Pie chart showing the distribution of karyotypes of 18 MDS/AML cases. Monosomy 7 abnormalities are listed on the left. (R) Somatic mutation profile of age-matched short telomere patients separated by those with (9 male/5 female) and without (11 male/9 female) MDS/AML. The age for each patient is included on top as is the presence of a monosomy 7 abnormality [−7, del(7q) or der(1;7)(q10;p10)]. All sequencing was performed on blood, except for the 2 AML cases (denoted as bolded ages) for which bone marrow was used. The ring sideroblasts (defined as >15% of cells examined) are denoted below the MDS/AML cases by an asterisk (*). Of note, another patient (of a total of 7) had ring sideroblasts but was not included in this analysis because of insufficient DNA. The respective germline mutations for each case are annotated in supplemental Figure 1A.
Short telomeres are sufficient to drive the somatic mutational landscape of MDS/AML
We analyzed the cytogenetic and genomic landscape of these short telomere myeloid cancers. All of the MDS/AML cases had abnormal karyotypes (100%). The most common abnormality was monosomy 7, alone or with other aberrations (56%; 10 of 17 with available data) (Figure 2Q; Table 3). We also centrally performed next-generation sequencing and found that 79% of patients (11 of 14) carried at least 1 somatic change in an MDS/AML gene (mean VAF, 12.6%; range, 2.17% to 35.3%; Figure 2R; supplemental Table 4). This mutation prevalence and the mutant genes mirror what is seen in unselected MDS/AML cohorts.40,41 We noted a trend toward mutual exclusivity of monosomy 7 with mutant epigenetic regulators (Figure 2R), but the immediate significance of this finding is unclear. These data indicated that although telomere-mediated MDS/AML may have some distinguishing clinical-pathologic features, its somatic genetic landscape overlaps with unselected MDS/AML.
A subset of MDS/AML has a recognizable granulocyte telomere attrition pattern by flowFISH
Because telomere shortening has been linked to genome instability in some settings, we tested whether patients who developed MDS/AML had shorter telomeres. We found 2 notable patterns by flowFISH analysis. First, short telomere MDS/AML patients had longer lymphocyte telomere length than short telomere aplastic anemia patients (mean difference from age-adjusted median, −2.5 vs −4.3 kb, respectively; P < .001; Mann-Whitney U test; Figure 3A). MDS/AML patients were also on average >3 decades older at diagnosis than those with aplastic anemia (median age at diagnosis, 54.0 vs 21.5 years; P < .001; Mann-Whitney U test; Figure 3B). Second, when we compared short telomere MDS/AML telomere lengths to age-matched short telomere patients who did not have MDS/AML, we saw no differences in lymphocyte measurements (Figure 3C). However, there was a marked discordance with granulocyte telomeres showing significant shortening compared with age-matched short telomere patients from our same cohort who did not have MDS/AML (lymphocyte-to-granulocyte difference in mean Δ telomere length, −0.9 kb vs −0.1 kb; P = .001; Wilcoxon-Matched paired nonparametric test; Figure 3C). Moreover, although none of the 20 non-MDS/AML short telomere patients had discordance (more than −1.0 kb), 8 of 17 MDS/AML patients (47%) showed this relative granulocyte shortening (P < .001; Fisher exact test). This myeloid attrition was acquired, as evidenced in the example of 1 pulmonary fibrosis patient who developed coincident discordance at the time of MDS diagnosis (Figure 3D). We examined, but found no significant clinical or pathologic features, including cytogenetic abnormality, that distinguished these MDS/AML patients with discordance from those without discordance.
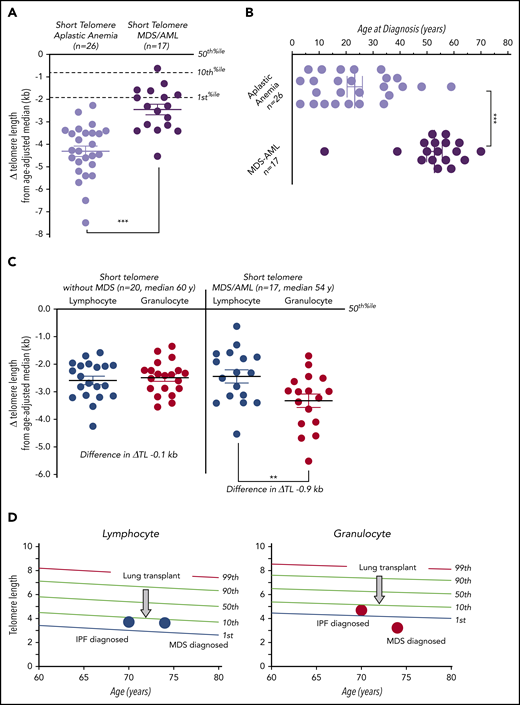
Telomere length discordance in short telomere patients with MDS/AML. (A) Mean of Δ telomere length from age-adjusted median in lymphocytes for patients with aplastic anemia and MDS/AML. (B) Age of diagnosis for patients with aplastic anemia and MDS/AML shows older age of onset for the latter group. (C) The degree of deviation from the age-adjusted median in lymphocytes and granulocytes in short telomere cases without (n = 20; median age, 60 years; range, 45-76 years; 13 male/7 female) and with MDS/AML (n = 17; median age, 54 years; range, 12-74 years; 11 male/6 female). Mean is graphed plus or minus standard error of the mean. **P = .001 (Mann-Whitney U test); ***P < .001 (Wilcoxon rank matched-pair test). (D) Longitudinal lymphocyte and granulocyte telomere lengths for 1 individual obtained at idiopathic pulmonary fibrosis (IPF) diagnosis and 4 years later at MDS diagnosis. The intervening age of lung transplant is graphically annotated by the arrow at age 72.
Telomere length discordance in short telomere patients with MDS/AML. (A) Mean of Δ telomere length from age-adjusted median in lymphocytes for patients with aplastic anemia and MDS/AML. (B) Age of diagnosis for patients with aplastic anemia and MDS/AML shows older age of onset for the latter group. (C) The degree of deviation from the age-adjusted median in lymphocytes and granulocytes in short telomere cases without (n = 20; median age, 60 years; range, 45-76 years; 13 male/7 female) and with MDS/AML (n = 17; median age, 54 years; range, 12-74 years; 11 male/6 female). Mean is graphed plus or minus standard error of the mean. **P = .001 (Mann-Whitney U test); ***P < .001 (Wilcoxon rank matched-pair test). (D) Longitudinal lymphocyte and granulocyte telomere lengths for 1 individual obtained at idiopathic pulmonary fibrosis (IPF) diagnosis and 4 years later at MDS diagnosis. The intervening age of lung transplant is graphically annotated by the arrow at age 72.
High rates of CHIP-associated mutations in adult short telomere syndrome patients
Because telomere-related MDS/AML showed a similar mutational landscape to age-related disease, we tested whether adult short telomere mutation carriers without MDS/AML have a higher rate of clonal hematopoiesis associated with aging and with the entity of clonal hematopoiesis of indeterminate potential (CHIP). We used the same targeted next-generation sequencing panel to assess 20 individuals who were age-matched to the MDS/AML patients (55% male; median age, 59 years; range, 45-76 years; Figure 2R). We found that 30% (6 of 20) had at least 1 CHIP-associated mutation defined by a VAF ≥2% threshold (Figure 2R; Table 4); this threshold has been used to quantify risk for myeloid cancer development in prior studies.42,43 The mean VAF was 8.2% (range, 2.2% to 32.9%; supplemental Table 5). This clonal hematopoiesis prevalence is higher than the 6% to 10% estimates seen in significantly older cohorts of healthy controls older than 70 years of age using the same VAF ≥2% threshold.42,43 One patient with a germline TR mutation had 3 independent TET2 loss-of-function clonal mutations detected, supporting a strong advantage under the pressure of short telomere hematopoiesis (Table 4). Three other short telomere patients had somatic TP53 mutations (mean VAF, 6.1%; range, 2.5% to 8.6%); all 3 died of pulmonary fibrosis within 4 years and none developed MDS/AML during the follow-up interval (Table 4). None of the 6 individuals with CHIP mutations showed the discordance patterns we saw in telomere-related MDS/AML (supplemental Figure 1B). These data indicated that although clonal hematopoiesis–associated mutations are common in adult short telomere patients, they are not sufficient to drive the granulocyte telomere attrition seen in one-half of MDS/AML patients with overt malignant disease.
Somatic mutations and follow-up of short telomere syndrome patients with CHIP-associated mutations (n = 6)
| Age, y | M/F | Germline mutation | Somatic mutation(s) | Somatic VAF, % | Marrow failure | Follow-up from NGS, mo | Survival |
|---|---|---|---|---|---|---|---|
| 52 | F | TR r.80U>A | GNAS Ile119_Glu122del | 32.9 | Moderate AA | 6 | Died, IPF |
| TET2 Gln275Ilefs*18 | 6.2 | ||||||
| TET2 Ala1341Cysfs*3 | 5.4 | ||||||
| TET2 Pro1115Leufs*2 | 3.3 | ||||||
| 54 | F | RTEL1 c.1135+1G>A | CBLB c.1594-1G>T | 7.0 | ↑MCV | 32 | Alive, PF |
| 55 | F | RTEL1 Arg986Ter | TP53 Arg282Trp | 2.5 | None | 34 | Died, lung transplant* |
| 66 | M | Unknown (low TR) | TP53 Gly245Ser | 8.6 | ↑MCV, mild thrombocytopenia | 7 | Died, IPF |
| U2AF1 Ser34Phe | 5.8 | ||||||
| ETV6 Leu376Pro | 5.4 | ||||||
| TET2 Gln810Ter | 2.2 | ||||||
| 67 | M | TERT Arg858Trp | TP53 Leu265Pro | 7.2 | None | 40 | Died, PF |
| 76 | M | Unknown | ASXL1 Gly646Trpfs*12 | 12.3 | None | 20 | Alive, IPF |
| Age, y | M/F | Germline mutation | Somatic mutation(s) | Somatic VAF, % | Marrow failure | Follow-up from NGS, mo | Survival |
|---|---|---|---|---|---|---|---|
| 52 | F | TR r.80U>A | GNAS Ile119_Glu122del | 32.9 | Moderate AA | 6 | Died, IPF |
| TET2 Gln275Ilefs*18 | 6.2 | ||||||
| TET2 Ala1341Cysfs*3 | 5.4 | ||||||
| TET2 Pro1115Leufs*2 | 3.3 | ||||||
| 54 | F | RTEL1 c.1135+1G>A | CBLB c.1594-1G>T | 7.0 | ↑MCV | 32 | Alive, PF |
| 55 | F | RTEL1 Arg986Ter | TP53 Arg282Trp | 2.5 | None | 34 | Died, lung transplant* |
| 66 | M | Unknown (low TR) | TP53 Gly245Ser | 8.6 | ↑MCV, mild thrombocytopenia | 7 | Died, IPF |
| U2AF1 Ser34Phe | 5.8 | ||||||
| ETV6 Leu376Pro | 5.4 | ||||||
| TET2 Gln810Ter | 2.2 | ||||||
| 67 | M | TERT Arg858Trp | TP53 Leu265Pro | 7.2 | None | 40 | Died, PF |
| 76 | M | Unknown | ASXL1 Gly646Trpfs*12 | 12.3 | None | 20 | Alive, IPF |
AA, aplastic anemia; IPF, idiopathic pulmonary fibrosis; MCV, mean corpuscular volume; PF, pulmonary fibrosis; NGS, next-generation sequencing.
This patient died of extrahematopoietic complications including renal failure 30 months after lung transplantation.
Short telomere comorbidities complicate the course of MDS/AML
We finally assessed the natural history of telomere-mediated MDS/AML (Figure 4). One- and 2-year survival rates were 61% and 39%, respectively, and median survival from the time of diagnosis was 15 months (range, 1-69 months) (Figure 4). At last follow-up, only 4 of 18 MDS/AML patients were alive (median, 51 months; range, 39-66 months) and all 4 surviving patients had undergone organ transplantation. Two underwent HSCT and 2 were diagnosed with indolent MDS prior to or after lung transplantation (Figure 4). In only rare cases (21%; 3 of 14) was the cause of death directly related to refractory MDS/AML or its hematologic complications. In the vast majority (65%; 9 of 14), extrahematopoietic short telomere syndrome diagnoses (pulmonary fibrosis [n = 7] or hepatopulmonary syndrome [n = 2]) were the primary causes of death. Two other patients died of hematologic complications but in the setting of renal failure requiring renal replacement therapy. Although standard medial MDS therapies were attempted (ie, transfusions, erythropoietin-stimulating, hypomethylating agents), they did not seem to significantly alter the clinical course in patients who had end-stage lung, liver, or renal disease (Figure 4).
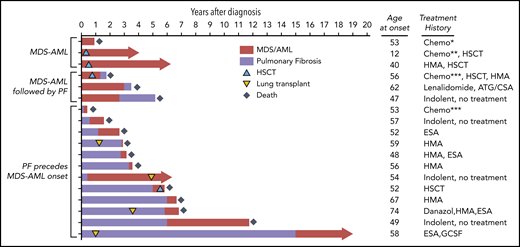
Treatment outcomes and clinical course of telomere-mediated MDS/AML. Swimmer plot of outcomes of 18 short telomere patients with MDS/AML clustered by co-occurrence of pulmonary fibrosis (PF). The age and treatment are shown to the right. Red bar shows time since diagnosis of MDS/AML, and purple indicates time since onset of pulmonary fibrosis symptoms. HSCT and lung transplant are graphically indicated along the timelines. The length of the bar represents follow-up through death or through last assessment. *This patient received standard acute lymphoblastic leukemia (ALL) therapy and therapy-related AML was diagnosed 8 months into the treatment, then he was treated with a mitoxantrone/etoposide salvage regimen. ***These 2 patients received standard intensive induction for high grade MDS and AML. ATG, antithymocyte globulin; Chemo, chemotherapy; CSA, cyclosporine; ESA, erythropoietin-stimulating agent; GCSF, granulocyte colony-stimulating factor; HMA, hypomethylating agent.
Treatment outcomes and clinical course of telomere-mediated MDS/AML. Swimmer plot of outcomes of 18 short telomere patients with MDS/AML clustered by co-occurrence of pulmonary fibrosis (PF). The age and treatment are shown to the right. Red bar shows time since diagnosis of MDS/AML, and purple indicates time since onset of pulmonary fibrosis symptoms. HSCT and lung transplant are graphically indicated along the timelines. The length of the bar represents follow-up through death or through last assessment. *This patient received standard acute lymphoblastic leukemia (ALL) therapy and therapy-related AML was diagnosed 8 months into the treatment, then he was treated with a mitoxantrone/etoposide salvage regimen. ***These 2 patients received standard intensive induction for high grade MDS and AML. ATG, antithymocyte globulin; Chemo, chemotherapy; CSA, cyclosporine; ESA, erythropoietin-stimulating agent; GCSF, granulocyte colony-stimulating factor; HMA, hypomethylating agent.
Discussion
The data we report here, based on a 16-year hospital-based experience, indicate that although increased over the general population, the cancer spectrum in the monogenic short telomere syndromes is narrow. Solid tumor prevalence is overall low, and the squamous cell mucosal phenotype is distinct from the common solid tumors associated with aging. Although short telomere patients had a significantly higher risk of developing MDS and AML, the overall incidence was still relatively low with these cancers affecting 16% of adults over the age of 50 years. Our data suggest there are 2 natural histories for cancer in the short telomere syndromes with solid tumors predominantly affecting younger male patients, especially those with classic dyskeratosis congenita, and MDS/AML predominantly affecting adults over the age of 50 years. The older age at diagnosis for MDS/AML raises a question as to the utility of regular “surveillance” marrows especially in children and young adults. We report here a recognizable pattern of myeloid-specific telomere attrition by flowFISH in peripheral blood; this was coincident with overt MDS/AML but is not sensitive as it was only identified in one-half of MDS/AML cases. This pattern nonetheless may be useful for assessing the possibility of disease progression in some patients using a blood-based assay. Prospective studies will be helpful in this area.
Short telomeres are the primary determinant of onset and severity of telomere-mediated disease,5,27 and, to date, there have been no clear telomere length–independent genotype-phenotype correlations. We report here an increased prevalence of squamous cancers, and likely cancer in general, in DKC1-mutant male patients with classic dyskeratosis congenita that is independent of telomere length. This finding, if replicated, could inform approaches to screening in this patient subset. DKC1 encodes the dyskerin protein, a pseudouridylase involved in the stability of telomerase RNA as well as hundreds of other box H/ACA RNAs including those involved in splicing and catalyzing ribosome RNA modification.44 Although telomerase RNA is sufficient to rescue the replicative defect of DKC1-mutant cells in vitro,45 the deficiency of other RNAs could contribute to cancer risk in vivo independent of replicative defects. Telomere length–independent factors, including impaired translation of tumor-suppressor transcripts, have been previously implicated in promoting tumorigenesis in a dyskerin hypomorphic mouse.46,47 Our data suggest that cancer-screening efforts may be higher yield if focused on subsets of male subjects with classic dyskeratosis congenita.
This is the first study, to our knowledge, to report on the somatic mutational landscape of cancer in the short telomere syndromes. Surprisingly, in the MDS/AML patients we studied, the somatic mutational landscape was similar to unselected MDS/AML. This observation raises the possibility that short telomeres may contribute to the biology of age-associated hypoplastic MDS/AML in some cases. We also found CHIP-associated mutations in one-third of short telomere syndrome adults who did not have MDS/AML. The recurrence of independent clonal mutations in CHIP-associated genes, including TET2, in some of these adults supports short telomere length being sufficient to select for age-related clonal hematopoiesis mutations. A prior study of children with classic dyskeratosis congenita reported evidence for clonal hematopoiesis but did not examine/report CHIP-associated changes.48 The mechanisms by which telomeres are maintained in patients with these myeloid cancers under replicative stress is not known. We did find that MDS/AML patients have significantly longer telomere length than children with aplastic anemia and are >3 decades older, and it is established that aplastic anemia is rare in adults with short telomere syndromes.27 These observations support a model wherein clonal evolution in the setting of “permissive” telomere capacity, and over decades, can evolve to malignancy, in contrast to aplastic anemia in which short telomere length is constraining. Clonal gain-of-function TERT promoter mutations have been reported in up to 10% of adults with telomerase mutations,49 although these patients did not have any myeloid malignancy. Overall, because age is the biggest risk factor for the acquisition of CHIP-related mutations, our data suggest a possible role for short telomere length as a driver of the CHIP signature with aging.
Novel germline variants will be submitted to the Telomerase Database (telomerase.asu.edu), and novel somatic variants will be submitted to The Catalogue Of Somatic Mutations In Cancer (cancer.sanger.ac.uk/cosmic).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to patients, study participants, and all of their referring clinicians. The authors appreciate helpful discussions with Jane Churpek. The authors acknowledge the support of the Johns Hopkins Genetic Resources Core Facility and Johns Hopkins Genomics staff.
This work was supported by National Institutes of Health grants RO1 CA225027 (National Cancer Institute) and RO1 HL119476 (National Heart, Lung, and Blood Institute), and the S&R, Gary Williams, and Commonwealth Foundations (M.A.). The authors would also like to acknowledge a gift in the name of Mrs P. Godrej (M.A.). K.E.S. was supported by National Heart, Lung, and Blood Institute grant T32HL007525 and the Turock Scholars Fund to the Telomere Center at Johns Hopkins. Johns Hopkins Genomics received funding from National Cancer Institute grant P30 CA006973.
Authorship
Contribution: K.E.S. and M.A. designed the study and reviewed and assembled all of the primary data; L.H. and C.D.G. performed the somatic-sequencing studies; A.L.B. designed and performed the SEER analyses; S.K.D. and A.E.D. analyzed some primary clinical data; A.S.D. reviewed and analyzed the hematopathology studies; M.A. wrote the paper with K.E.S.; and all of the authors reviewed and gave input on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mary Armanios, Johns Hopkins University School of Medicine, 1650 Orleans St, CRB 1 Room 186, Baltimore, MD 21287; e-mail: marmani1@jhmi.edu.
