

Abstract
Erdheim-Chester disease (ECD) is a rare histiocytosis that was recently recognized as a neoplastic disorder owing to the discovery of recurrent activating MAPK (RAS-RAF-MEK-ERK) pathway mutations. Typical findings of ECD include central diabetes insipidus, restrictive pericarditis, perinephric fibrosis, and sclerotic bone lesions. The histopathologic diagnosis of ECD is often challenging due to nonspecific inflammatory and fibrotic findings on histopathologic review of tissue specimens. Additionally, the association of ECD with unusual tissue tropism and an insidious onset often results in diagnostic errors and delays. Most patients with ECD require treatment, except for a minority of patients with minimally symptomatic single-organ disease. The first ECD consensus guidelines were published in 2014 on behalf of the physicians and researchers within the Erdheim-Chester Disease Global Alliance. With the recent molecular discoveries and the approval of the first targeted therapy (vemurafenib) for BRAF-V600–mutant ECD, there is a need for updated clinical practice guidelines to optimize the diagnosis and treatment of this disease. This document presents consensus recommendations that resulted from the International Medical Symposia on ECD in 2017 and 2019. Herein, we include the guidelines for the clinical, laboratory, histologic, and radiographic evaluation of ECD patients along with treatment recommendations based on our clinical experience and review of literature in the molecular era.
Introduction
Erdheim-Chester disease (ECD) is a rare histiocytic disorder with diverse clinical manifestations, ranging from indolent, localized presentations to life-threatening, multisystem disease. Since the first publication of ECD guidelines in 2014,1 new biological insights and regulatory advances, including recurrent somatic mutations in the MAPK pathway beyond BRAF-V600E2 and the approval of a targeted agent (vemurafenib) for BRAF-V600–mutant ECD3 in the United States, have transformed diagnosis and management approaches. Historically considered an inflammatory, nonneoplastic disorder, ECD is now included in the 2016 World Health Organization (WHO) classification of hematopoietic tumors4 and has been grouped among the “L” (Langerhans) group of the 2016 revised histiocytosis classification of the Histiocyte Society.5 The diagnosis and therapy of ECD have evolved and become increasingly complex, necessitating updated guidelines.
Methods
An international, multidisciplinary group of scientists and physicians engaged in ECD research and management convened at the annual ECD Medical Symposium on 24 October 2017, followed by 10 July 2019, to update ECD guidelines in light of (1) new scientific developments since 2014 and (2) the members’ evolving approach to ECD evaluation and treatment. The group was composed of experts from hematology, internal medicine, molecular biology, neurology, oncology, and pathology with extensive experience in the diagnosis and treatment of ECD and related histiocytic disorders. Collectively, the coauthors have been involved in the care of ∼800 ECD patients. An English-language search of PubMed and Web of Science was conducted for ECD-related literature from January 1996 until December 2019. The recommendations presented here emerged from literature review and expert opinion. Key recommendation statements (Table 1) generated from in-person discussion were evaluated individually by the coauthors and their agreement or disagreement was recorded. The degree of consensus for each statement is categorized into: A (strong consensus: ≥95%), B (consensus: 75% to 95%), and C (majority agreement: 50% to 75%). Statements that had ≤50% agreement are not presented.
Consensus recommendations for diagnosis and treatment of ECD
| Consensus recommendations | Category of consensus* |
|---|---|
| Diagnosis | |
| A biopsy of lesional tissue is strongly recommended even in circumstances of highly suggestive clinical and imaging features not only to confirm ECD diagnosis, but also to establish BRAF mutational status and perform sequencing for MAPK-ERK pathway and other somatic mutations | A |
| ECD without any bone lesions can occur (<10%) but should only be considered in the context of suggestive histopathology or highly characteristic nonosseous lesions (ie, perinephric stranding, periaortic infiltrates, right atrial pseudotumor, or a combination of these with or without central diabetes insipidus) and ideally with supportive mutational data (BRAF or MAPK-ERK pathway mutations) | A |
| ECD should still be considered in the presence of characteristic clinical/radiologic features, even when biopsy does not demonstrate classic xanthomatous histiocytes; meticulous osseous imaging for lesions in the tibia and femur are critical in such cases, as well as mutational analysis of tissue for BRAF and MAPK-ERK pathway mutations | A |
| Baseline full-body (vertex-to-toes) FDG-PET-CT, including the distal extremities, is recommended in all cases to aid in diagnosis and define the extent of disease; if FDG-PET cannot be performed, contrast-enhanced CT of the chest, abdomen, and pelvis can be performed as well as imaging of the lower extremities (CT, MRI, or 99Tc bone scan) | A |
| MRI of the brain with gadolinium is recommended in all patients at diagnosis | A |
| MRI of the heart is recommended in all patients at diagnosis; if an MRI cannot be performed, CT scan and/or echocardiogram should be performed | B |
| Laboratory studies are performed to assess for renal insufficiency, cytopenias, markers of inflammation (C-reactive protein), and evidence of endocrinopathy and anterior pituitary function | A |
| Due to a high prevalence of concomitant myeloid neoplasms in patients with ECD, bone marrow biopsy should be considered, especially in the context of otherwise unexplained cytopenias/cytosis or monocytosis | A |
| Confirmation of negative BRAF-V600E mutational testing using >1 genotyping modality and/or genotyping of biopsies from >1 anatomic site should be performed (particularly when lesions from bone are found to be BRAF wild type) | A |
| IHC for VE1 is not felt to be sensitive or specific as the sole method for BRAF-V600E mutational analysis and should be confirmed with another molecular assay if feasible | B |
| In the absence of sufficient tumor tissue, cfDNA analysis from peripheral blood can be used for assessment of BRAF-mutational status; however, the sensitivity of such assays may be variable | A |
| Treatment | |
| Treatment is indicated for most ECD patients, except some select cases of asymptomatic minimal burden disease, which can be monitored closely | B |
| Systemic corticosteroids, surgery, and radiation therapy may be used to relieve edema or acute symptoms, but are not recommended as monotherapies for ECD | A |
| For patients with BRAF-V600E ECD who have cardiac/neurologic disease or end-organ dysfunction, BRAF-inhibitor therapy with vemurafenib or dabrafenib should be implemented as first-line therapy | A |
| For BRAF-V600E ECD without end-organ dysfunction, BRAF-inhibitors or conventional therapy may both be considered for first-line therapy based on toxicity profile and drug availability/experience of clinician | A |
| For ECD patients without BRAF-V600E and cardiac/neurologic disease or end-organ dysfunction, empiric treatment with MEK-inhibitor should be strongly considered as first-line therapy | A |
| Optimal duration and dosing of targeted therapies is not known, although relapse has been observed in the majority of cases following complete cessation of BRAF-inhibitors; maintenance treatment in the setting of metabolic remission with low-dose therapy as tolerated may be considered | A |
| For patients without access to targeted therapies and high-burden disease, IFN-α/PEG–IFN-α or cladribine therapy may be considered | A |
| For patients with low-burden disease involving bones and retroperitoneum, cytokine-directed therapy such as anakinra may be appropriate first-line therapy | B |
| Response assessment and monitoring | |
| Full-body (vertex-to-toes) FDG-PET-CT should be performed every 2-6 mo after initiation of a new therapy for response assessment; once best response is established on 2 scans and disease is stabilized with steady dose of drug, the frequency of PET imaging can be individualized, ranging from every 6 mo to longer intervals | B |
| Organ-specific imaging of involved disease sites (CT or MRI) should be performed every 2-6 mo initially after beginning treatment of response assessment; once best response is established on 2 scans and disease is stabilized with steady dose of drug, the frequency of imaging can be individualized, ranging from every 6 mo to longer intervals; a separate CT may not be necessary if performed in conjunction with FDG-PET | B |
| Endocrinopathies persist or can develop despite treatment of ECD; therefore, annual endocrine evaluation is recommended | A |
| Treatment with targeted and immunosuppressive agents (including IFN-α/PEG–IFN-α) should be continued indefinitely if tolerated, however, attempting cessation of treatment or lowering of dose for patients with minimal or stable disease for a prolonged period of time may by reasonable on case-by-case basis | A |
| Consensus recommendations | Category of consensus* |
|---|---|
| Diagnosis | |
| A biopsy of lesional tissue is strongly recommended even in circumstances of highly suggestive clinical and imaging features not only to confirm ECD diagnosis, but also to establish BRAF mutational status and perform sequencing for MAPK-ERK pathway and other somatic mutations | A |
| ECD without any bone lesions can occur (<10%) but should only be considered in the context of suggestive histopathology or highly characteristic nonosseous lesions (ie, perinephric stranding, periaortic infiltrates, right atrial pseudotumor, or a combination of these with or without central diabetes insipidus) and ideally with supportive mutational data (BRAF or MAPK-ERK pathway mutations) | A |
| ECD should still be considered in the presence of characteristic clinical/radiologic features, even when biopsy does not demonstrate classic xanthomatous histiocytes; meticulous osseous imaging for lesions in the tibia and femur are critical in such cases, as well as mutational analysis of tissue for BRAF and MAPK-ERK pathway mutations | A |
| Baseline full-body (vertex-to-toes) FDG-PET-CT, including the distal extremities, is recommended in all cases to aid in diagnosis and define the extent of disease; if FDG-PET cannot be performed, contrast-enhanced CT of the chest, abdomen, and pelvis can be performed as well as imaging of the lower extremities (CT, MRI, or 99Tc bone scan) | A |
| MRI of the brain with gadolinium is recommended in all patients at diagnosis | A |
| MRI of the heart is recommended in all patients at diagnosis; if an MRI cannot be performed, CT scan and/or echocardiogram should be performed | B |
| Laboratory studies are performed to assess for renal insufficiency, cytopenias, markers of inflammation (C-reactive protein), and evidence of endocrinopathy and anterior pituitary function | A |
| Due to a high prevalence of concomitant myeloid neoplasms in patients with ECD, bone marrow biopsy should be considered, especially in the context of otherwise unexplained cytopenias/cytosis or monocytosis | A |
| Confirmation of negative BRAF-V600E mutational testing using >1 genotyping modality and/or genotyping of biopsies from >1 anatomic site should be performed (particularly when lesions from bone are found to be BRAF wild type) | A |
| IHC for VE1 is not felt to be sensitive or specific as the sole method for BRAF-V600E mutational analysis and should be confirmed with another molecular assay if feasible | B |
| In the absence of sufficient tumor tissue, cfDNA analysis from peripheral blood can be used for assessment of BRAF-mutational status; however, the sensitivity of such assays may be variable | A |
| Treatment | |
| Treatment is indicated for most ECD patients, except some select cases of asymptomatic minimal burden disease, which can be monitored closely | B |
| Systemic corticosteroids, surgery, and radiation therapy may be used to relieve edema or acute symptoms, but are not recommended as monotherapies for ECD | A |
| For patients with BRAF-V600E ECD who have cardiac/neurologic disease or end-organ dysfunction, BRAF-inhibitor therapy with vemurafenib or dabrafenib should be implemented as first-line therapy | A |
| For BRAF-V600E ECD without end-organ dysfunction, BRAF-inhibitors or conventional therapy may both be considered for first-line therapy based on toxicity profile and drug availability/experience of clinician | A |
| For ECD patients without BRAF-V600E and cardiac/neurologic disease or end-organ dysfunction, empiric treatment with MEK-inhibitor should be strongly considered as first-line therapy | A |
| Optimal duration and dosing of targeted therapies is not known, although relapse has been observed in the majority of cases following complete cessation of BRAF-inhibitors; maintenance treatment in the setting of metabolic remission with low-dose therapy as tolerated may be considered | A |
| For patients without access to targeted therapies and high-burden disease, IFN-α/PEG–IFN-α or cladribine therapy may be considered | A |
| For patients with low-burden disease involving bones and retroperitoneum, cytokine-directed therapy such as anakinra may be appropriate first-line therapy | B |
| Response assessment and monitoring | |
| Full-body (vertex-to-toes) FDG-PET-CT should be performed every 2-6 mo after initiation of a new therapy for response assessment; once best response is established on 2 scans and disease is stabilized with steady dose of drug, the frequency of PET imaging can be individualized, ranging from every 6 mo to longer intervals | B |
| Organ-specific imaging of involved disease sites (CT or MRI) should be performed every 2-6 mo initially after beginning treatment of response assessment; once best response is established on 2 scans and disease is stabilized with steady dose of drug, the frequency of imaging can be individualized, ranging from every 6 mo to longer intervals; a separate CT may not be necessary if performed in conjunction with FDG-PET | B |
| Endocrinopathies persist or can develop despite treatment of ECD; therefore, annual endocrine evaluation is recommended | A |
| Treatment with targeted and immunosuppressive agents (including IFN-α/PEG–IFN-α) should be continued indefinitely if tolerated, however, attempting cessation of treatment or lowering of dose for patients with minimal or stable disease for a prolonged period of time may by reasonable on case-by-case basis | A |
cfDNA, cell-free DNA; CT, computed tomography; FDG, 18F-fluoro-deoxyglucose; IFN-α, interferon-α; IHC, immunohistochemistry; MRI, magnetic resonance imaging; PEG-IFN-α, pegylated interferon-α.
A (strong consensus: ≥95%), B (consensus: 75% to 95%).
Epidemiology
Although ∼800 cases of ECD have been reported in the literature, the exact incidence is unknown given the lack of population-based mandatory reporting to national registries. Epidemiologic data on ECD in the United Stated are derived from a prospective natural history study of 60 patients by the National Institutes of Health (NIH) and in Europe from a French cohort of 165 patients. ECD is primarily a disease of middle-aged adults, with a mean age of 46 years at diagnosis in the United States (range, 20-74 years)6 and 56 years in the French cohort (range, 29-86 years).7 ECD was noted in both studies to have a male preponderance (70% to 75%). Pediatric ECD cases are exceptional but do exist, and may present initially with a central nervous system juvenile xanthogranuloma (JXG) mass lesion, frequently harboring BRAF-V600E, also with a male preponderance.8
Molecular pathogenesis and cell of origin
Molecular pathogenesis of ECD and alterations beyond BRAF-V600E
It was previously unclear whether ECD was a benign or malignant disorder, in part due to difficulty in establishing clonality and identifying driver mutations. Since 2012, a series of recurrent activating kinase mutations and fusions involving the canonical MAPK (RAS-RAF-MEK-ERK) and phosphatidylinositol 3-kinase (PI3K)-AKT pathways have been discovered in a large proportion of ECD patients (Figure 1).2,6,9-22 This discovery provided firm evidence that ECD is a clonal neoplastic disorder driven by constitutive MAPK signaling in most cases, and provided one of the first targets for molecular therapeutics in histiocytosis.13 In 2015, whole-exome sequencing on 14 ECD fresh-frozen specimens identified point mutations in the ARAF, MAP2K1, NRAS, and PI3KCA genes.2 This study also found novel MAP2K1 mutations in 14% of cases (2 of 14). An additional 9 activating MAP2K1 mutations were discovered in 50% of BRAF–wild-type, archived ECD cases (9 of 18) evaluated by targeted sequencing in a validation cohort in this study.2 These MAP2K1 mutations have been demonstrated in vitro to cause constitutive activation of MEK1.2 Although MAP2K1 mutations can be seen in ECD, they are not exclusive to histiocytosis and can be seen in other hematopoietic neoplasms as well. Other mutations in ECD include activating mutations in NRAS and KRAS, which have been described in 4 independent studies.10,12,14,20 PI3K-AKT pathway alterations (PI3KCA mutations) have been reported in 2 studies.2,12 Mutations in ARAF were found in 21% of ECD specimens (3 of 14) with 2 of them being mutually exclusive of BRAF-V600E.2 Functionally activating gene fusions involving the ALK gene have also been reported in ECD.2,23 Interestingly, similar MAPK–extracellular signal-regulated kinase (ERK) pathway mutations have been recently reported in 57% to 100% of histiocytic sarcoma specimens, suggesting a potential correlation between the 2 neoplasms.24,25
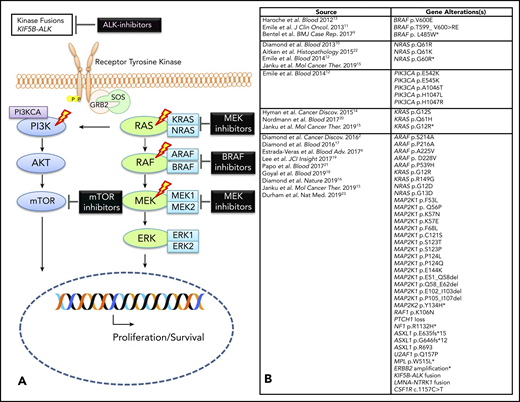
Molecular alterations in ECD. (A) Graphic depicting MAPK pathway signaling in ECD with therapeutic targets. The lightning bolts indicate the most common genes that are altered in ECD. (B) Composite of all somatic alterations (mutations and fusions) that have been reported in ECD to date. *Mutations identified in plasma cell-free DNA analysis only; therefore, these could reflect somatic mutations from another diagnostic entity (eg, clonal hematopoiesis).
Molecular alterations in ECD. (A) Graphic depicting MAPK pathway signaling in ECD with therapeutic targets. The lightning bolts indicate the most common genes that are altered in ECD. (B) Composite of all somatic alterations (mutations and fusions) that have been reported in ECD to date. *Mutations identified in plasma cell-free DNA analysis only; therefore, these could reflect somatic mutations from another diagnostic entity (eg, clonal hematopoiesis).
Histogenesis of ECD
Gene-set enrichment analysis of RNA-sequencing data from Langerhans cell histiocytosis (LCH) and non-LCH neoplastic cells suggests that neoplasms such as ECD have transcriptional profiles similar to myeloid progenitor cells or macrophages.2 Recently, rare novel activating mutations in CSF1R have been described in ECD and related histiocytoses, suggesting that a subset of these may possibly arise from the extraembryonic macrophage progenitors.23 Research into the ontogeny of BRAF-V600E ECD and LCH has identified mutant alleles in CD34+ hematopoietic stem/progenitor cells, monocytes, and myeloid dendritic cells, suggesting several potential precursor pathways for these neoplasms.26,27 Further studies into the role of other MAPK pathway–activating mutations and gene fusions on the histogenesis of ECD using preclinical models are needed to advance our understanding of the developmental dysregulation that leads to the pathological formation of histiocytic neoplasms, including the recruitment of nonclonal “bystander” immune cells.
Mixed histiocytosis: an overlap syndrome
New insights have been recently provided into the association of ECD with other histiocytic neoplasms, especially LCH.28 This entity, called “mixed histiocytosis,” was initially reported in individual case reports but was later confirmed in a French series of 23 patients.28 In these cases, the ECD component was either diagnosed subsequently or concomitantly with LCH, but never preceded it. These patients were found to be younger, with a higher frequency of BRAF-V600E mutations in the LCH (69%) and ECD (82%) lesions, as compared with the incidence reported for either entity alone (50% to 60%)28 Recently, ECD has been observed in association with the extranodal form of Rosai-Dorfman-Destombes disease (RDD), occurring predominantly in men, and frequently harboring MAP2K1 mutations.29 It is important for clinicians to be aware of the co-occurrence of these histiocytic neoplasms so that atypical manifestations alert them toward consideration of another biopsy to confirm overlapping entities.
Clinical and radiographic features
ECD has diverse organ system manifestations, with varying frequencies based on different institutional case series (Figures 2-4; Table 2).30-43 It is crucial for clinicians to be familiar with the manifestations due to their implications in diagnosis and prognostic staging of the disease. It is important to rule out concomitant myeloid neoplasms among ECD patients due to their high rates of co-occurrence.21,44

Diverse manifestations of ECD. (A) Coronal postcontrast chest computed tomography (CT) demonstrates extensive soft tissue sheathing of the thoracic aorta. (B) Enhancing lesions in the hypothalamic pituitary axis (HPA), brainstem, and cerebellar peduncle is shown in sagittal gadolinium enhanced T1 magnetic resonance imaging (MRI). (C) Three-dimensional fast imaging using steady-state acquisition (3D-FIESTA) MRI of the heart showing right atrial mass from ECD. (D) Maximal intensity projection (MIP) of 18F-fluoro-deoxyglucose (FDG)–positron emission tomography (PET) demonstrates typical hypermetabolic ECD lesions throughout the appendicular skeleton with greatest activity of the disease in the legs. (E) Irregular bilateral enhancing of ECD lesions in the middle cerebellar peduncles are demonstrated by postgadolinium axial T1 MRI. (F) Expansile irregularly enhancing ECD lesions in the pons seen on postgadolinium axial T1 MRI. (G) MIP of FDG-PET demonstrating ECD lesions with increased uptake in distal femur, orbit, multilevel thoracolumbar spine roots, and right atrium. (H) Periorbital xanthelasmas from ECD. (I-J) “Hairy kidney” hypermetabolic and contrast-enhancing perinephric infiltrates are shown on axial-fused FDG PET-CT and contrast-enhanced axial CT scan. (K) High-resolution axial CT scan image of the chest demonstrating reticulonodular opacities from ECD. (L) Atrophic or neurodegenerative changes in the brainstem and cerebellum are shown by axial T2-FLAIR MRI.
Diverse manifestations of ECD. (A) Coronal postcontrast chest computed tomography (CT) demonstrates extensive soft tissue sheathing of the thoracic aorta. (B) Enhancing lesions in the hypothalamic pituitary axis (HPA), brainstem, and cerebellar peduncle is shown in sagittal gadolinium enhanced T1 magnetic resonance imaging (MRI). (C) Three-dimensional fast imaging using steady-state acquisition (3D-FIESTA) MRI of the heart showing right atrial mass from ECD. (D) Maximal intensity projection (MIP) of 18F-fluoro-deoxyglucose (FDG)–positron emission tomography (PET) demonstrates typical hypermetabolic ECD lesions throughout the appendicular skeleton with greatest activity of the disease in the legs. (E) Irregular bilateral enhancing of ECD lesions in the middle cerebellar peduncles are demonstrated by postgadolinium axial T1 MRI. (F) Expansile irregularly enhancing ECD lesions in the pons seen on postgadolinium axial T1 MRI. (G) MIP of FDG-PET demonstrating ECD lesions with increased uptake in distal femur, orbit, multilevel thoracolumbar spine roots, and right atrium. (H) Periorbital xanthelasmas from ECD. (I-J) “Hairy kidney” hypermetabolic and contrast-enhancing perinephric infiltrates are shown on axial-fused FDG PET-CT and contrast-enhanced axial CT scan. (K) High-resolution axial CT scan image of the chest demonstrating reticulonodular opacities from ECD. (L) Atrophic or neurodegenerative changes in the brainstem and cerebellum are shown by axial T2-FLAIR MRI.
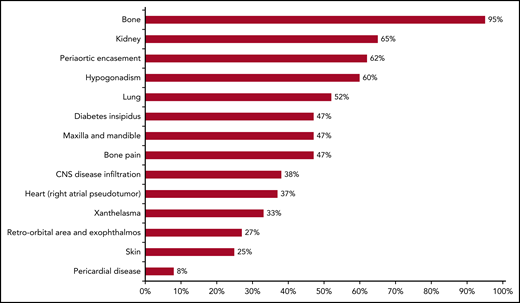
Frequency of clinical and radiological manifestations of ECD. Based on a prospective cohort study by Estrada-Veras et al.6 Each bar reflects the proportion of patients with reported findings mentioned on the y-axis.
Frequency of clinical and radiological manifestations of ECD. Based on a prospective cohort study by Estrada-Veras et al.6 Each bar reflects the proportion of patients with reported findings mentioned on the y-axis.
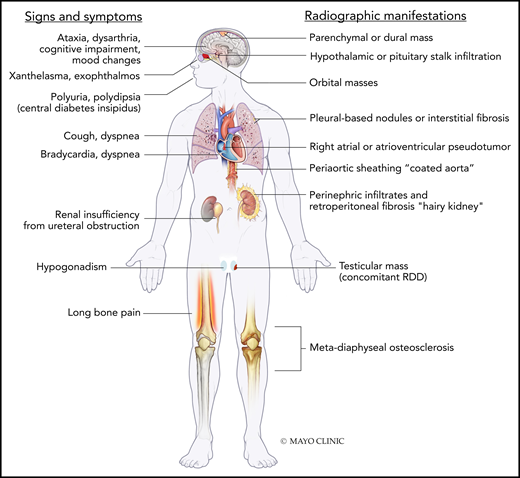
Signs/symptoms and radiographic features of ECD. RDD, Rosai-Dorfman-Destombes disease. Used with permission of Mayo Foundation for Medical Education and Research, all rights reserved.
Signs/symptoms and radiographic features of ECD. RDD, Rosai-Dorfman-Destombes disease. Used with permission of Mayo Foundation for Medical Education and Research, all rights reserved.
Summary of organ manifestations of ECD
| Organ | Clinical and radiographic features |
|---|---|
| Bones | Lower-extremity bone pain is the most common presenting symptom (50%)6 ; full-body (skull-to-toes) FDG-PET-CT scan or 99mTc bone scintigraphy shows bilateral symmetric osteosclerosis of metadiaphysis of femur, tibia, and fibula in >95% cases and is pathognomonic; skull and axial skeleton less commonly involved than LCH, which typically shows lytic punched-out lesions rather than sclerotic lesions that are seen in ECD30 |
| Nervous system | Seen in 25% to 50% patients, with predilection toward brain parenchyma of the posterior cranial fossa and dura, although infiltrations can occur throughout the neuraxis39 ; these abnormalities often but do not invariably demonstrate gadolinium enhancement; rarely, may present with nontumorous neurodegenerative-like (atrophic) changes in the brainstem and cerebellum Retinal involvement has been reported as well96 Clinical manifestations depend on the site of involvement, and may include ataxia, dysarthria, cognitive impairment, headaches, or peripheral neuropathy6 ; some patients may present with mood lability111 |
| Endocrine | Anterior and posterior pituitary abnormalities (40% to 70%); most commonly central DI (25% to 50%) and may precede the diagnosis of ECD by several years, sometimes decades6,35,39,109 ; other pituitary hormone deficiencies that are commonly seen are growth hormone, gonadotropin, thyrotropin, and corticotropin; hyperprolactinemia can be seen in 15% to 30% patients35,109 ; although normal pituitary imaging does not exclude hormonal deficiency, an abnormal pituitary stalk MRI is commonly associated with hypopituitarism; primary hypothyroidism (20%), hypogonadism (19%), and adrenal insufficiency (6%) can be seen as well35 |
| Orbit | Unilateral or bilateral infiltration seen in 25% to 30% of patients; clinical manifestations include exophthalmos, retro-orbital pain, oculomotor nerve palsy or vision loss6,36 ; differential diagnosis includes xanthogranuloma,36 RDD, Graves disease, granulomatous disease, and IgG4-related disease |
| Respiratory | Lung involvement in ECD is mostly asymptomatic and seen radiologically in ∼50% of cases, involving either the lung parenchyma or pleura6,7 ; unlike pulmonary LCH, no association with cigarette smoking has been reported; although plain chest radiographs can be normal, findings on CT of the chest may include mediastinal infiltration, pleural thickening or effusions, interlobular septal thickening, ground-glass opacities, or centrilobular opacities34,38,41 ; pulmonary function tests commonly reveal a more restrictive as compared with an obstructive pattern6,34 ; ECD may also involve facial sinuses, with maxillary sinus thickening in ∼50% patients6 |
| Cardiovascular | Cardiovascular involvement may be asymptomatic but can be seen in 50% to 70% patients at imaging evaluation by CT and/or MRI31,33,75 ; most common findings include pericardial infiltration with effusion (which may be complicated by tamponade) or myocardial infiltration in the form of right atrioventricular pseudotumor (40%)6,75 ; circumferential soft tissue sheathing of the thoracic/abdominal aorta and its branches may be seen as “coated” aorta may be seen on CT scan (50% to 60%)6,42 ; periarterial involvement of renal vessels may lead to renovascular hypertension (20%), is responsive to stenting,112 and can be monitored with renal artery dopplers; involvement of other visceral vessels has been reported as well; coronary arteries may be involved in 30% to 50% of patients31,38 |
| Retroperitoneum, including kidney | Manifesting as infiltrative perinephric soft tissue thickening, or “hairy kidney” (50% to 60%)7 ; perinephric infiltrates can rarely extend to involve the renal pelvis and/or renal ureters causing hydronephrosis and renal failure requiring dialysis and nephrostomy with stent placement6 ; in some cases, it may also extend to involve the adrenal glands32 ; longstanding perinephric ECD may lead to atrophy of kidneys; differential diagnosis includes retroperitoneal fibrosis and IgG4-related disease |
| Cutaneous | Seen in 20% to 30% patients, with one-half of the patients manifesting skin lesions as initial ECD presentation6,7,37 ; most common cutaneous manifestation is xanthelasma, occurring as yellowish plaques around the eyelids but can occur as yellowish-brown papules or plaques on the face, neck, axilla, trunk or groin as well37 ; it may also present as subcutaneous nodules or granuloma annulare-like lesions37 |
| Reticuloendothelial and hematopoietic system | In one study, 11% of ECD patients had liver and spleen involvement, respectively, although the prevalence has been lower in other series6,40 ; ECD rarely involves the lymph nodes, but may involve bone marrow in 8% of cases113 ; ECD may also be associated with concomitant myeloid neoplasms in 10% of cases, specifically myeloproliferative neoplasm, myelodysplastic syndrome, or mixed myelodysplastic/myeloproliferative overlap syndrome including chronic myelomonocytic leukemia21 |
| Organ | Clinical and radiographic features |
|---|---|
| Bones | Lower-extremity bone pain is the most common presenting symptom (50%)6 ; full-body (skull-to-toes) FDG-PET-CT scan or 99mTc bone scintigraphy shows bilateral symmetric osteosclerosis of metadiaphysis of femur, tibia, and fibula in >95% cases and is pathognomonic; skull and axial skeleton less commonly involved than LCH, which typically shows lytic punched-out lesions rather than sclerotic lesions that are seen in ECD30 |
| Nervous system | Seen in 25% to 50% patients, with predilection toward brain parenchyma of the posterior cranial fossa and dura, although infiltrations can occur throughout the neuraxis39 ; these abnormalities often but do not invariably demonstrate gadolinium enhancement; rarely, may present with nontumorous neurodegenerative-like (atrophic) changes in the brainstem and cerebellum Retinal involvement has been reported as well96 Clinical manifestations depend on the site of involvement, and may include ataxia, dysarthria, cognitive impairment, headaches, or peripheral neuropathy6 ; some patients may present with mood lability111 |
| Endocrine | Anterior and posterior pituitary abnormalities (40% to 70%); most commonly central DI (25% to 50%) and may precede the diagnosis of ECD by several years, sometimes decades6,35,39,109 ; other pituitary hormone deficiencies that are commonly seen are growth hormone, gonadotropin, thyrotropin, and corticotropin; hyperprolactinemia can be seen in 15% to 30% patients35,109 ; although normal pituitary imaging does not exclude hormonal deficiency, an abnormal pituitary stalk MRI is commonly associated with hypopituitarism; primary hypothyroidism (20%), hypogonadism (19%), and adrenal insufficiency (6%) can be seen as well35 |
| Orbit | Unilateral or bilateral infiltration seen in 25% to 30% of patients; clinical manifestations include exophthalmos, retro-orbital pain, oculomotor nerve palsy or vision loss6,36 ; differential diagnosis includes xanthogranuloma,36 RDD, Graves disease, granulomatous disease, and IgG4-related disease |
| Respiratory | Lung involvement in ECD is mostly asymptomatic and seen radiologically in ∼50% of cases, involving either the lung parenchyma or pleura6,7 ; unlike pulmonary LCH, no association with cigarette smoking has been reported; although plain chest radiographs can be normal, findings on CT of the chest may include mediastinal infiltration, pleural thickening or effusions, interlobular septal thickening, ground-glass opacities, or centrilobular opacities34,38,41 ; pulmonary function tests commonly reveal a more restrictive as compared with an obstructive pattern6,34 ; ECD may also involve facial sinuses, with maxillary sinus thickening in ∼50% patients6 |
| Cardiovascular | Cardiovascular involvement may be asymptomatic but can be seen in 50% to 70% patients at imaging evaluation by CT and/or MRI31,33,75 ; most common findings include pericardial infiltration with effusion (which may be complicated by tamponade) or myocardial infiltration in the form of right atrioventricular pseudotumor (40%)6,75 ; circumferential soft tissue sheathing of the thoracic/abdominal aorta and its branches may be seen as “coated” aorta may be seen on CT scan (50% to 60%)6,42 ; periarterial involvement of renal vessels may lead to renovascular hypertension (20%), is responsive to stenting,112 and can be monitored with renal artery dopplers; involvement of other visceral vessels has been reported as well; coronary arteries may be involved in 30% to 50% of patients31,38 |
| Retroperitoneum, including kidney | Manifesting as infiltrative perinephric soft tissue thickening, or “hairy kidney” (50% to 60%)7 ; perinephric infiltrates can rarely extend to involve the renal pelvis and/or renal ureters causing hydronephrosis and renal failure requiring dialysis and nephrostomy with stent placement6 ; in some cases, it may also extend to involve the adrenal glands32 ; longstanding perinephric ECD may lead to atrophy of kidneys; differential diagnosis includes retroperitoneal fibrosis and IgG4-related disease |
| Cutaneous | Seen in 20% to 30% patients, with one-half of the patients manifesting skin lesions as initial ECD presentation6,7,37 ; most common cutaneous manifestation is xanthelasma, occurring as yellowish plaques around the eyelids but can occur as yellowish-brown papules or plaques on the face, neck, axilla, trunk or groin as well37 ; it may also present as subcutaneous nodules or granuloma annulare-like lesions37 |
| Reticuloendothelial and hematopoietic system | In one study, 11% of ECD patients had liver and spleen involvement, respectively, although the prevalence has been lower in other series6,40 ; ECD rarely involves the lymph nodes, but may involve bone marrow in 8% of cases113 ; ECD may also be associated with concomitant myeloid neoplasms in 10% of cases, specifically myeloproliferative neoplasm, myelodysplastic syndrome, or mixed myelodysplastic/myeloproliferative overlap syndrome including chronic myelomonocytic leukemia21 |
DI, diabetes insipidus; IgG4, immunoglobulin G4.
Histopathologic features
Although classic histopathologic findings include foamy histiocytes with small nuclei and surrounding fibrosis, multinucleated giant cells, and Touton giant cells, ECD has a spectrum of histopathological features within the xanthogranuloma family, often in a milieu of reactive lymphocytes, plasma cells, and rarely neutrophils.45 ECD may also display atypical features such as florid lymphohistiocytic infiltrates or fibrotic lamellae with only scattered foamy histiocytes and rare/absent Touton giant cells.46 On immunohistochemistry (IHC), histiocytes are positive for CD68, CD163, factor XIIIa, and fascin, and negative for CD1a and CD207 (langerin). Although ECD was classically thought to be negative for S100 by IHC, weak or focally positive staining has been observed in 20% to 30% cases (Table 3).6,46 Although it is important to differentiate ECD from other histiocytic disorders for appropriate diagnosis, as well as to identify overlapping entities (Table 3), absence of the classical description of foamy histiocytes and Touton cells does not preclude a diagnosis of ECD.
Summary of the pathological, molecular, and radiological features of the histiocytic disorders
| Disease | ECD | JXG/AXG | ALK+ histiocytosis | RDD | LCH |
|---|---|---|---|---|---|
| Pathologic features | |||||
| Xanthomatous histiocytes | Yes | Yes | Variable | No | No |
| Touton giant cells | Yes (mainly dermal sites) | Yes (mainly dermal sites) | Rarely | No | No |
| Emperipolesis (intracytoplasmic inflammatory cells including plasma cells and lymphocytes) | Rare | Rare | Rare | Abundant | No |
| Nuclear features | Bland; round-to-oval; small; no grooves | Bland; round-to-oval; small; no grooves | Bland; round-to-oval; small; typically no grooves | Large round; hypochromatic | Oval; retiform irregular nuclear contours |
| Nucleoli | Inconspicuous | Inconspicuous | Inconspicuous | Variable inconspicuous to distinct | Inconspicuous |
| Cytoplasm | Classically abundant, xanthomatous but often overlap with JXG/AXG | Compact; pink; glassy; progressively xanthomatous | Abundant; eosinophilic; typically not xanthomatous | Abundant foamy, clear without xanthomatous features; frequent emperipolesis | Abundant; eosinophilic |
| Immunophenotype | |||||
| CD68 (cytoplasmic) | ++ | ++ | ++ | ++ | + (paranuclear cytoplasmic dot) |
| CD163 (surface) | ++ | ++ | ++ | ++ | — |
| CD14 (surface) | ++ | ++ | ++ | ++ | — |
| CD1a (surface) | − | − | − | − | ++ |
| CD207 (Langerin) (cytoplasmic) | − | − | − | − | ++ |
| S100 (cytoplasmic/nuclear) | −/+ (light) | −/+ (light) | −/++ (in some cases dark staining) | + | + |
| Factor XIIIa (cytoplasmic) | + | + | + | + | − |
| Fascin (cytoplasmic) | + | + | + | + | − |
| CD45 (light surface) | + | + | + | + | + |
| BRAF VE1 (cytoplasmic) | ++* | − (Positive cases should be strongly favored to be in ECD family) | − | − (Rare case reports ++) | ++* |
| ALK (cytoplasmic) | ++* | ++* | ++* | − | − |
| NTRK1(cytoplasmic) | ++* | ++* | − | − | − |
| Molecular features | |||||
| BRAF V600E | Frequent (50%) | Reported (3%) | No | Reported (3%) | Frequent (55%) |
| MAP2K1 | Common (18%) | Common (12%) | No | Common (15%) | Common (15%) |
| RAS isoforms (KRAS, NRAS) | Common (8%) | Common (10%) | No | Common (30%) | Rare (2%) |
| BRAF deletions | Rare (2%) | No | No | No | Common (6%) |
| PI3K isoforms (PIK3CA, PIK3CD) | Reported (3%) | Rare (1%) | No | No | Rare (1%) |
| ARAF | Reported (4%) | Rare (1%) | No | Reported (3%) | Rare (1%) |
| Other BRAF missense | No | No | No | No | Reported (3%) |
| RAF1 | Rare (1%) | No | No | No | No |
| MAP2K2 | Rare (1%) | No | No | No | No |
| MAP3K1 | Reported (1 case) (Amplification) | No | No | No | Reported |
| CSF1R | Rare (1%) | Common (10%) | No | Rare (1%) | Rare (1%) |
| BRAF fusions | Rare (2%) | Common (6%) | No | No | Reported (3%) |
| ALK fusions | Reported (3%) | Reported (3%) | Frequent (100%) | No | No |
| NTRK1 fusions | Rare (1%) | Common (10%) | No | No | No |
| RET fusions | No | Reported (3%) | No | No | No |
| ETV3-NCOA2 fusion | No | No | No | No | Rare (1%) |
| Disease | ECD | JXG/AXG | ALK+ histiocytosis | RDD | LCH |
|---|---|---|---|---|---|
| Pathologic features | |||||
| Xanthomatous histiocytes | Yes | Yes | Variable | No | No |
| Touton giant cells | Yes (mainly dermal sites) | Yes (mainly dermal sites) | Rarely | No | No |
| Emperipolesis (intracytoplasmic inflammatory cells including plasma cells and lymphocytes) | Rare | Rare | Rare | Abundant | No |
| Nuclear features | Bland; round-to-oval; small; no grooves | Bland; round-to-oval; small; no grooves | Bland; round-to-oval; small; typically no grooves | Large round; hypochromatic | Oval; retiform irregular nuclear contours |
| Nucleoli | Inconspicuous | Inconspicuous | Inconspicuous | Variable inconspicuous to distinct | Inconspicuous |
| Cytoplasm | Classically abundant, xanthomatous but often overlap with JXG/AXG | Compact; pink; glassy; progressively xanthomatous | Abundant; eosinophilic; typically not xanthomatous | Abundant foamy, clear without xanthomatous features; frequent emperipolesis | Abundant; eosinophilic |
| Immunophenotype | |||||
| CD68 (cytoplasmic) | ++ | ++ | ++ | ++ | + (paranuclear cytoplasmic dot) |
| CD163 (surface) | ++ | ++ | ++ | ++ | — |
| CD14 (surface) | ++ | ++ | ++ | ++ | — |
| CD1a (surface) | − | − | − | − | ++ |
| CD207 (Langerin) (cytoplasmic) | − | − | − | − | ++ |
| S100 (cytoplasmic/nuclear) | −/+ (light) | −/+ (light) | −/++ (in some cases dark staining) | + | + |
| Factor XIIIa (cytoplasmic) | + | + | + | + | − |
| Fascin (cytoplasmic) | + | + | + | + | − |
| CD45 (light surface) | + | + | + | + | + |
| BRAF VE1 (cytoplasmic) | ++* | − (Positive cases should be strongly favored to be in ECD family) | − | − (Rare case reports ++) | ++* |
| ALK (cytoplasmic) | ++* | ++* | ++* | − | − |
| NTRK1(cytoplasmic) | ++* | ++* | − | − | − |
| Molecular features | |||||
| BRAF V600E | Frequent (50%) | Reported (3%) | No | Reported (3%) | Frequent (55%) |
| MAP2K1 | Common (18%) | Common (12%) | No | Common (15%) | Common (15%) |
| RAS isoforms (KRAS, NRAS) | Common (8%) | Common (10%) | No | Common (30%) | Rare (2%) |
| BRAF deletions | Rare (2%) | No | No | No | Common (6%) |
| PI3K isoforms (PIK3CA, PIK3CD) | Reported (3%) | Rare (1%) | No | No | Rare (1%) |
| ARAF | Reported (4%) | Rare (1%) | No | Reported (3%) | Rare (1%) |
| Other BRAF missense | No | No | No | No | Reported (3%) |
| RAF1 | Rare (1%) | No | No | No | No |
| MAP2K2 | Rare (1%) | No | No | No | No |
| MAP3K1 | Reported (1 case) (Amplification) | No | No | No | Reported |
| CSF1R | Rare (1%) | Common (10%) | No | Rare (1%) | Rare (1%) |
| BRAF fusions | Rare (2%) | Common (6%) | No | No | Reported (3%) |
| ALK fusions | Reported (3%) | Reported (3%) | Frequent (100%) | No | No |
| NTRK1 fusions | Rare (1%) | Common (10%) | No | No | No |
| RET fusions | No | Reported (3%) | No | No | No |
| ETV3-NCOA2 fusion | No | No | No | No | Rare (1%) |
Immunophenotype key: −, negative; +, weak positive; ++, moderate to strong positive.
AXG, adult xanthogranuloma.
Moderate to strong positivity should correlate with molecular alteration; BRAF VE1, ALK and pTRK are mutually exclusive.
Diagnosis
The rarity of ECD, coupled with its protean characteristics, can make the diagnosis extremely challenging and often requires integration of descriptive pathology together with clinical and radiographic findings. ECD patients may see multiple providers and undergo several biopsies that have, historically, led to delayed diagnosis and institution of therapy in most patients, with the average time from symptom onset to diagnosis being a few months to several years.6,47 One scenario leading to delayed diagnosis occurs when histopathological features of the biopsy material are suggestive of, but not considered diagnostic for ECD because of the absence of classical ECD morphology as discussed in “Histopathologic features.” ECD is not exclusively a pathologic diagnosis, and it is necessary to interpret histopathologic features in conjunction with clinical, radiographic, and, as of recently, molecular findings.
A defining feature of ECD is symmetric osteosclerosis of the metadiaphysis of the lower-extremity bones on studies such as plain radiographs, 99mTc bone scintigraphy, 18F-fluoro-deoxyglucose (FDG)–positron emission tomography (PET), computed tomography (CT), or magnetic resonance imaging (MRI) (Figure 2). Although bone scintigraphy is the most sensitive of these for detecting osseous lesions, FDG PET-CT is preferred as a diagnostic test by virtue of its ability to assess other organ involvement.48,49 It must be noted that it is crucial to obtain the PET-CT scan as a full-body (skull-to-toes) test, as compared with skull-to-mid-thigh, as the latter may not capture these characteristic osseous lesions. Previously proposed diagnostic criteria required presence of both osteosclerotic lesions in the legs.1,50 However, a small proportion of ECD (∼5%) may not demonstrate long-bone involvement6 and in such cases the diagnosis hinges on other features.
Role of molecular testing in ECD diagnosis
As most ECD patients harbor activating somatic mutations or fusions in the genes of the MAPK-ERK or the PI3K-AKT pathway, molecular profiling of biopsy material can increase confidence in an ECD diagnosis in cases with ambiguous histopathological findings and/or absence of osseous lesions. It is notable that tissue genotyping may not uncover a driver alteration in a small proportion of patients (10% to 15%)51 or there may be insufficient cellularity in the specimen to conduct molecular analysis. In such cases, a properly validated BRAF-VE1 or phosphorylated ERK stain may help if there is moderate to strong cytoplasmic staining in the lesional cells (Table 1).
Baseline evaluation and molecular assessment of tissue
Baseline evaluation
The goal of the evaluation in newly diagnosed patients is to define the extent of disease involvement, assess subsequent risk of end-organ compromise, and define a plan of treatment and surveillance (Figure 5). Regardless of symptoms, we recommend FDG PET-CT imaging including the brain and distal extremities, MRI of the brain with gadolinium, and cardiac MRI in all newly diagnosed patients. Even in cases in which 99mTc bone scanning has been performed initially for diagnostic purposes, FDG PET-CT is recommended for initial evaluation to assess organ involvement and as a tool for guiding biopsy targets.52-54 Dedicated CT of the chest, abdomen, and pelvis is recommended to demonstrate pulmonary, periaortic, and perinephric infiltrates. In some cases, further organ-specific imaging may be necessary based on clinical and radiologic findings to better characterize the involvement of certain sites. Additionally, laboratory studies are needed to assess endocrinopathies, peripheral blood count abnormalities, renal/hepatic function, immunological assessment, and the degree of inflammation.
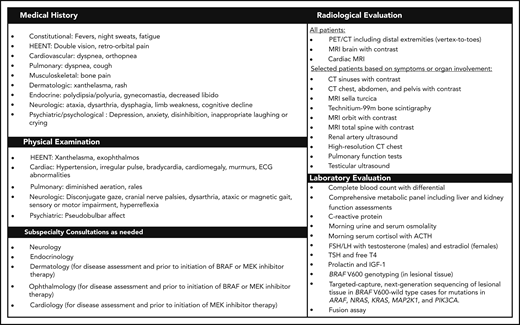
Suggested workup for a newly diagnosed or suspected patient with ECD. ACTH, adrenocorticotropic hormone; ECG, electrocardiogram; FSH/LH, follicle-stimulating hormone/luteinizing hormone; HEENT, head, eyes, ears, nose, and throat; IGF-1, insulin-like growth factor 1; TSH, thyroid-stimulating hormone.
Suggested workup for a newly diagnosed or suspected patient with ECD. ACTH, adrenocorticotropic hormone; ECG, electrocardiogram; FSH/LH, follicle-stimulating hormone/luteinizing hormone; HEENT, head, eyes, ears, nose, and throat; IGF-1, insulin-like growth factor 1; TSH, thyroid-stimulating hormone.
Tissue biopsy and histopathologic assessment
Tissue biopsy is required in all ECD cases, not only for confirmation of diagnosis, but also to allow identification of associated mutations for therapeutic purposes. The selection of the biopsy site for specimen acquisition can be challenging in ECD due to low tumor cellularity and heterogeneity of lesions. The diagnosis is often made by biopsy of one of the skin, osseous, or soft tissue perinephric infiltrates/lesions using a percutaneous CT-guided approach. If an FDG PET-CT has been performed prior to biopsy, we recommend a biopsy of the most FDG-avid sites that are accessible and safe, especially in cases of bony lesions. Because of the variable components of histiocytic infiltrate and surrounding stroma, multiple core biopsies are recommended to optimize the yield of tissue for histopathologic review and molecular testing. Biopsy of xanthelasmas or other skin lesions, if present, offers a less-invasive alternative.46 If DNA-based testing is planned, it is important to coordinate tissue handling of bone biopsies because standard decalcification of bone samples will lead to the destruction of informative DNA. Alternatively, an EDTA-based decalcification method can be used, which can help preserve DNA integrity.
Molecular assessment of tissue for alterations in MAPK-ERK and other pathways
BRAF-V600E mutation testing should be pursued for all patients. There are several methods to test for this mutation, including IHC, polymerase chain reaction (PCR), pyrosequencing, droplet digital PCR (ddPCR), and targeted-capture next-generation sequencing (NGS). Although IHC is a cost-efficient and reliable method of testing for the BRAF-V600E protein in other cancers, the authors’ shared experience is that this method is not as sensitive for the evaluation of ECD material, similar to previous reports in LCH.55 Hence, all negative or equivocal IHC tests should be confirmed by a sensitive sequencing technique on the same or alternative tissue specimens. Although not available at most clinical laboratories, ddPCR is the most sensitive method, and, in many cases, BRAF-V600E is present at remarkably low allele fractions (<5%).56 Clinical presentations with cerebral, cardiac, and orbital disease may prompt more exhaustive testing for BRAF-V600E because the incidence of this mutation is highest in these phenotypes.7
In cases without the BRAF-V600E mutation, we recommend targeted-capture NGS with a commercially available assay to test alterations in other genes of the MAPK-ERK and PI3K-AKT pathways (KRAS, NRAS, ARAF, RAF1, MAP2K1, MAP2K2, BRAF indels, and PI3KCA). Of note, ∼40% of BRAF-V600–wild-type patients will harbor a mutation in MAP2K1. Most of these panels also include RNA sequencing that tests known oncogenic kinase fusions that have been reported previously in ECD (Figure 1). Data regarding concordance between tumor-based sequencing and cell-free DNA (cfDNA)-based sequencing have varied, with high concordance in BRAF-V600E mutant cases and low in others.14,15,57 In cases for which a tissue specimen is insufficient for molecular analysis, cfDNA testing is a reasonable alternative.
Treatment
Most patients with ECD require systemic treatment at diagnosis, with the exception of asymptomatic nonvital single-organ (eg, bone) or minimally symptomatic disease that may be monitored. The therapeutic landscape of ECD and related histiocytic neoplasms has changed drastically over the last 5 years due to the discovery of activating and targetable MAPK-ERK pathway mutations in the vast majority of patients. In the following sections (Table 4), we summarize the various treatment options for ECD. Where possible, clinical trial enrollment for ECD patients is encouraged in order to establish new treatment options.
Treatments and clinical trials for ECD
| Class of treatment Medication | Dose and schedule | Comment |
|---|---|---|
| BRAF inhibitors | ||
| Vemurafenib | 480-960 mg twice daily | Nearly 100% metabolic response in several case series, and in 1 prospective clinical trial3,59,60,62-66 US-FDA approval in November 2017; start with 240-480 mg twice daily and modify based on observed response and toxicities; most common adverse effects include cutaneous complications (rash, squamous cell cancer), arthralgia, QTc prolongation, and fatigue; pretreatment dermatology examination and electrocardiogram; monitoring with electrocardiogram every 3 mo and semiannual dermatology examination |
| Dabrafenib | 75-150 mg twice daily | Successful treatment reported in several case reports and 1 series20,61,67 ; anecdotal experience reflects similar efficacy to vemurafenib, and less cutaneous toxicity than vemurafenib; start with 50-75 mg twice daily and modify based on observed response and toxicities; pretreatment dermatology examination and electrocardiogram; monitoring every 3 mo with electrocardiogram and semiannual dermatology examination |
| MEK inhibitors | ||
| Cobimetinib | 20-60 mg daily for 21 of 28-d cycle | 3 published cases/series and a prospective clinical trial of responses to single-agent cobimetinib therapy in BRAF-V600E and BRAF-V600–wild type2,67,72,114 ; notable toxicities include serous retinopathy (reversible), rash, cardiomyopathy, and rarely rhabdomyolysis; pretreatment echocardiogram, ophthalmologic, and dermatologic evaluation; recheck ophthalmologic examination 2-3 wk after initiation and then every 3-4 mo for the first year of treatment; monitoring every 3 mo with echocardiogram and semiannual dermatology examination |
| Trametinib | 1-2 mg daily | Two cases of response to single-agent trametinib for ECD with KRAS,20 MAP2K1,2 and NRAS mutations21 ; anecdotal experience reflects similar efficacy and toxicity to cobimetinib; pretreatment echocardiogram, ophthalmologic, and dermatologic evaluation; recheck ophthalmologic examination 2-3 wk after initiation; monitoring every 3 monthly echocardiogram and semiannual dermatology examination |
| Combined BRAF and MEK inhibitors | ||
| Vemurafenib + cobimetinib or dabrafenib + trametinib | Doses similar as above | Case reports of combination therapy with dabrafenib and trametinib with robust responses in BRAF-V600-ECD6,73 ; Similar anecdotal experience with vemurafenib and cobimetinib; may consider in rare instances of suboptimal response to BRAF-inhibitor or toxicity necessitating dose reduction |
| First-line conventional therapy | ||
| PEG-IFN-α | 135 μg SC/wk (standard dose) or 180 μg SC/wk (high dose) | Currently, the conventional therapy with largest evidence-base in ECD79-84 ; case series have demonstrated survival benefit with the use of some form of IFN-α; high-dose IFN-α for patients with CNS or cardiac involvement; major limitation is the high frequency of systemic adverse effects |
| IFN-α | 3 mIU SC TIW (standard dose) or 6-9 mIU SC TIW (high dose) | Currently, the conventional therapy with largest evidence-base in ECD79-84 ; case series have demonstrated survival benefit with the use of some form of IFN-α; high-dose IFN-α for patients with CNS or cardiac involvement; major limitation is the high frequency of systemic adverse effects |
| Cladribine | 5 mg/m2 IV daily for 5 d Q4 wk 0.10 μg/kg SC daily for 5 d Q4 wk | 3 published cases and a retrospective series of 21 patients treated with cladribine was published demonstrating ∼50% clinical or radiologic response rate94,95,98 ; prophylactic antimicrobials against Pneumocystis jirovecii (cotrimoxazole) and viruses (acyclovir, valacyclovir) should be added during the duration of the treatment and until the lymphocyte count normalize |
| Anakinra | 100 mg SC daily or up to 2 mg/kg/d | Several case reports of successful treatment, mainly of less severe forms of ECD,86,87 but limited reports of CNS17 or cardiac87 disease with favorable response; especially effective for bone pain and constitutional symptoms; injection site reactions may be seen occasionally but otherwise well tolerated |
| Second-line conventional therapy | ||
| Sirolimus and prednisone | Sirolimus dosed to level of 8-12 ng/mL | 8 of 10 patients had a favorable response in a prospective clinical trial75 ; only one-third of patients with CNS or bone disease had a response |
| Imatinib | 400 mg PO daily | Mixed results in 7 ECD patients treated with imatinib76,77 ; lack of efficacy in several other cases anecdotally |
| Infliximab | 5 mg/mg IV Q6 wk | 2 patients with cardiac disease refractory to treatment with IFN-α had clinical improvement when treated infliximab,91 and another case series of 12 patients showed ∼40% response rates mainly cardiac and cerebellar92 |
| Tocilizumab | 8 mg/kg IV Q4 wk or 162 mg SC weekly | 2 of 3 patients in a pilot phase 2 trial had a favorable response, both without CNS involvement93 ; may be considered in patients with bone only or cardiac disease |
| Methotrexate | 7.5-25 mg PO or SC/wk | 3 of 13 patients had a partial response in a case series, ongoing responses seen in conjunctival and choroidal disease107 ; may be considered in ocular disease |
| High-dose methotrexate (3.5 mg/m2 IV) | One case of response to high-dose methotrexate103 | |
| Clinical trials (experimental) | ||
| Dabrafenib + trametinib | Dabrafenib 150 mg twice daily | Phase 2; NCT03794297; BRAF-V600–mutant ECD without prior BRAF-inhibitor or MEK-inhibitor therapy |
| Trametinib 2 mg once daily | ||
| PLX8394 (BRAF-inhibitor) | Dose escalation study | Phase 1/2; NCT02428712; previously treated BRAF mutated |
| Bevacizumab (VEGF-inhibitor) and Temsirolimus (mTOR-inhibitor) alone or combination with valproic acid or cetuximab (EGFR-inhibitor) | Dose escalation study | Phase 1/2; NCT01552434; newly diagnosed or previously treated |
| Cobimetinib (MEK-inhibitor) | 60 mg oral daily for days 1-21 of each 28-d cycle | Phase 2; NCT02649972; newly diagnosed or previously treated; interim results show promising activity in ECD patients114 |
| Cobimetinib (MEK-inhibitor) | 40 mg oral daily for days 1-21 of each 28-d cycle | Phase 2: NCT04007848; newly diagnosed or previously treated BRAF–wild-type histiocytosis, 2:1 randomized trial with placebo control |
| Cobimetinib (MEK-inhibitor) | 60 mg oral daily for days 1-21 of each 28-d cycle | Phase 2; NCT04079179; newly diagnosed or previously treated histiocytosis; pediatric and adult patients |
| Class of treatment Medication | Dose and schedule | Comment |
|---|---|---|
| BRAF inhibitors | ||
| Vemurafenib | 480-960 mg twice daily | Nearly 100% metabolic response in several case series, and in 1 prospective clinical trial3,59,60,62-66 US-FDA approval in November 2017; start with 240-480 mg twice daily and modify based on observed response and toxicities; most common adverse effects include cutaneous complications (rash, squamous cell cancer), arthralgia, QTc prolongation, and fatigue; pretreatment dermatology examination and electrocardiogram; monitoring with electrocardiogram every 3 mo and semiannual dermatology examination |
| Dabrafenib | 75-150 mg twice daily | Successful treatment reported in several case reports and 1 series20,61,67 ; anecdotal experience reflects similar efficacy to vemurafenib, and less cutaneous toxicity than vemurafenib; start with 50-75 mg twice daily and modify based on observed response and toxicities; pretreatment dermatology examination and electrocardiogram; monitoring every 3 mo with electrocardiogram and semiannual dermatology examination |
| MEK inhibitors | ||
| Cobimetinib | 20-60 mg daily for 21 of 28-d cycle | 3 published cases/series and a prospective clinical trial of responses to single-agent cobimetinib therapy in BRAF-V600E and BRAF-V600–wild type2,67,72,114 ; notable toxicities include serous retinopathy (reversible), rash, cardiomyopathy, and rarely rhabdomyolysis; pretreatment echocardiogram, ophthalmologic, and dermatologic evaluation; recheck ophthalmologic examination 2-3 wk after initiation and then every 3-4 mo for the first year of treatment; monitoring every 3 mo with echocardiogram and semiannual dermatology examination |
| Trametinib | 1-2 mg daily | Two cases of response to single-agent trametinib for ECD with KRAS,20 MAP2K1,2 and NRAS mutations21 ; anecdotal experience reflects similar efficacy and toxicity to cobimetinib; pretreatment echocardiogram, ophthalmologic, and dermatologic evaluation; recheck ophthalmologic examination 2-3 wk after initiation; monitoring every 3 monthly echocardiogram and semiannual dermatology examination |
| Combined BRAF and MEK inhibitors | ||
| Vemurafenib + cobimetinib or dabrafenib + trametinib | Doses similar as above | Case reports of combination therapy with dabrafenib and trametinib with robust responses in BRAF-V600-ECD6,73 ; Similar anecdotal experience with vemurafenib and cobimetinib; may consider in rare instances of suboptimal response to BRAF-inhibitor or toxicity necessitating dose reduction |
| First-line conventional therapy | ||
| PEG-IFN-α | 135 μg SC/wk (standard dose) or 180 μg SC/wk (high dose) | Currently, the conventional therapy with largest evidence-base in ECD79-84 ; case series have demonstrated survival benefit with the use of some form of IFN-α; high-dose IFN-α for patients with CNS or cardiac involvement; major limitation is the high frequency of systemic adverse effects |
| IFN-α | 3 mIU SC TIW (standard dose) or 6-9 mIU SC TIW (high dose) | Currently, the conventional therapy with largest evidence-base in ECD79-84 ; case series have demonstrated survival benefit with the use of some form of IFN-α; high-dose IFN-α for patients with CNS or cardiac involvement; major limitation is the high frequency of systemic adverse effects |
| Cladribine | 5 mg/m2 IV daily for 5 d Q4 wk 0.10 μg/kg SC daily for 5 d Q4 wk | 3 published cases and a retrospective series of 21 patients treated with cladribine was published demonstrating ∼50% clinical or radiologic response rate94,95,98 ; prophylactic antimicrobials against Pneumocystis jirovecii (cotrimoxazole) and viruses (acyclovir, valacyclovir) should be added during the duration of the treatment and until the lymphocyte count normalize |
| Anakinra | 100 mg SC daily or up to 2 mg/kg/d | Several case reports of successful treatment, mainly of less severe forms of ECD,86,87 but limited reports of CNS17 or cardiac87 disease with favorable response; especially effective for bone pain and constitutional symptoms; injection site reactions may be seen occasionally but otherwise well tolerated |
| Second-line conventional therapy | ||
| Sirolimus and prednisone | Sirolimus dosed to level of 8-12 ng/mL | 8 of 10 patients had a favorable response in a prospective clinical trial75 ; only one-third of patients with CNS or bone disease had a response |
| Imatinib | 400 mg PO daily | Mixed results in 7 ECD patients treated with imatinib76,77 ; lack of efficacy in several other cases anecdotally |
| Infliximab | 5 mg/mg IV Q6 wk | 2 patients with cardiac disease refractory to treatment with IFN-α had clinical improvement when treated infliximab,91 and another case series of 12 patients showed ∼40% response rates mainly cardiac and cerebellar92 |
| Tocilizumab | 8 mg/kg IV Q4 wk or 162 mg SC weekly | 2 of 3 patients in a pilot phase 2 trial had a favorable response, both without CNS involvement93 ; may be considered in patients with bone only or cardiac disease |
| Methotrexate | 7.5-25 mg PO or SC/wk | 3 of 13 patients had a partial response in a case series, ongoing responses seen in conjunctival and choroidal disease107 ; may be considered in ocular disease |
| High-dose methotrexate (3.5 mg/m2 IV) | One case of response to high-dose methotrexate103 | |
| Clinical trials (experimental) | ||
| Dabrafenib + trametinib | Dabrafenib 150 mg twice daily | Phase 2; NCT03794297; BRAF-V600–mutant ECD without prior BRAF-inhibitor or MEK-inhibitor therapy |
| Trametinib 2 mg once daily | ||
| PLX8394 (BRAF-inhibitor) | Dose escalation study | Phase 1/2; NCT02428712; previously treated BRAF mutated |
| Bevacizumab (VEGF-inhibitor) and Temsirolimus (mTOR-inhibitor) alone or combination with valproic acid or cetuximab (EGFR-inhibitor) | Dose escalation study | Phase 1/2; NCT01552434; newly diagnosed or previously treated |
| Cobimetinib (MEK-inhibitor) | 60 mg oral daily for days 1-21 of each 28-d cycle | Phase 2; NCT02649972; newly diagnosed or previously treated; interim results show promising activity in ECD patients114 |
| Cobimetinib (MEK-inhibitor) | 40 mg oral daily for days 1-21 of each 28-d cycle | Phase 2: NCT04007848; newly diagnosed or previously treated BRAF–wild-type histiocytosis, 2:1 randomized trial with placebo control |
| Cobimetinib (MEK-inhibitor) | 60 mg oral daily for days 1-21 of each 28-d cycle | Phase 2; NCT04079179; newly diagnosed or previously treated histiocytosis; pediatric and adult patients |
EGFR, epidermal growth factor receptor; Q4, every 4; Q6, every 6; PO, postoperatively; SC, subcutaneously; TIW, 3 times a week; VEGF, vascular endothelial growth factor.
Targeted therapies
BRAF-inhibitors (vemurafenib, dabrafenib, encorafenib)
Vemurafenib is approved by the US Food and Drug Administration for BRAF-V600–mutant ECD based on the results of a phase II trial,58 which demonstrated a 62% response rate by Response evaluation criteria in solid tumors (RECIST) criteria and 100% response rate by FDG-PET-CT in 22 patients.3 Most patients in this study required a dose reduction to 480 mg twice daily due to intolerance, which is our recommended initial dose. The findings of this trial were consistent with several reports of the efficacy of BRAF inhibitors, notably from the French cohort at the Pitié-Salpêtrière hospital.59-66 Vemurafenib often leads to dramatic and rapid responses in all disease sites, and has led to the reversal of critical illness from ECD in some cases.64 Although BRAF-inhibitor therapy generally achieves robust and durable responses, a study from the French cohort demonstrated that 75% of patients who discontinued vemurafenib relapsed within 6 months.67 Rescue treatment with a BRAF inhibitor, however, recaptured responses in all patients. A notable limitation of this study was that several patients only received BRAF inhibitors for a few months and discontinued without achieving a complete remission (CR). BRAF-inhibitors paradoxically also increase the risk of secondary neoplasia presumably by activation of RAS signaling in BRAF–wild-type cells.21,68,69 Such activation may also lead to rare adverse effects such as sarcoidosis and pancreatitis.70,71 A careful discussion of the risks and benefits of ongoing treatment is warranted. Resistance to BRAF-inhibitors is unusual in histiocytic disorders, with only a single report of a BRAF-V600E ECD patient developing a new KRAS-mutant lesion after treatment with dabrafenib.20 Future studies are needed to ascertain stringent definition of CR (depth and duration) to better inform drug discontinuation.
MEK-inhibitors (cobimetinib, trametinib, binimetinib, selumetinib)
The accumulating evidence for the existence of other activating MAPK-ERK pathway mutations (ARAF, KRAS, MAP2K1) among non-BRAF-V600–ECD led to an interest in the exploration of downstream blockade of this pathway using MEK inhibitors. This approach was used successfully in 5 refractory ECD patients without BRAF-V600E mutation who had a robust response to either cobimetinib or trametinib.2,72 These results led to 2 phase 2 clinical trials of cobimetinib in patients with histiocytic disorders (Table 4). The first planned analysis of this study showed an 89% overall response rate (ORR) by FDG-PET-CT in histiocytosis patients without the BRAF-V600E mutations or those who could not tolerate BRAF inhibitors due to toxicity.16 Responses were seen irrespective of the sites of disease. Similar efficacy has also been reported with trametinib in ECD patients without BRAF-V600E mutations.15,20 There are currently no data regarding cessation of MEK-inhibitor therapy and frequency or timing of subsequent relapse.
Combination of BRAF- and MEK-inhibitors
Similar to melanoma, combination approaches using BRAF and MEK inhibitors have been used successfully in ECD, and there is an ongoing clinical trial (NCT03794297).67,73 However, unlike melanoma, ECD is quite sensitive to kinase-inhibitor monotherapy and the combination may not provide higher response rates. Although combination therapy has demonstrated a lower incidence of cutaneous toxicities than BRAF-inhibitor alone in melanoma studies, some other toxicities (fatigue, arthralgia, cardiac failure) may be additive in nature.74 Hence, combination therapy use should probably be limited to cases of suboptimal response to BRAF-inhibitors alone or unmanageable cutaneous toxicities.
mTOR inhibitors (sirolimus, everolimus)
Eleven percent to 17% of ECD patients demonstrate activation of the mammalian target of rapamycin (mTOR) pathway through PIK3CA mutations, which can potentially be blocked using mTOR inhibitors.2,12 A phase 2 trial used sirolimus with prednisone in 10 ECD patients and resulted in an ORR of 80% in at least 1 disease site.75 Responses were seen in 50% of retroperitoneal and 75% of cardiovascular lesions, but no patients achieved CR. None of the 5 patients who were tested showed the presence of PI3KCA mutations, but mTOR pathway activation was demonstrated in tissues using IHC. mTOR inhibitors are not recommended as first-line therapy for ECD treatment but may be a therapeutic option in refractory ECD patients.
Other tyrosine kinase inhibitors (imatinib, sorafenib)
One of 7 patients treated with imatinib had a favorable response.76,77 One clinical and radiographic response to treatment was observed with the multikinase inhibitor, sorafenib, in refractory ECD with ARAF-S214A mutation. Outside of clinical trials of novel rational agents, the promising activity of MEK-inhibitors has eclipsed the role of other kinase inhibitors for management of ECD.
Other potential targeted agents
With the recent discovery of CSF1R mutations, ALK and RET fusions, there may be a role for targeted agents beyond BRAF- and MEK-inhibitors in ECD and other histiocytic neoplasms. Indeed, there are reports of successful treatment with crizotinib in an ECD patient harboring a KIF5B-ALK fusion and with selpercatinib in a disseminated JXG patient with a NCOA4-RET fusion.23 Other potential therapeutic targets may include ERK inhibitors due to the constitutive MAPK-ERK activation in ECD or pexidartinib among patients with CSF1R mutations.
Conventional (immunosuppressive or chemotherapeutic) therapies
IFN-α and PEG–IFN-α
Interferon-α-2a (IFN-α) and pegylated interferon-α-2a (PEG–IFN-α) are treatments with extensive experience in ECD, leading to response rates of 50% to 80%.7,78-84 In a case series of patients with cardiac and central nervous system (CNS) disease, higher doses (≥9 mIU of IFN-α or ≥180 μg of PEG–IFN-α) were needed to achieve a better response when lower doses were not sufficient, although most patients achieved a partial response or stable disease (64% CNS and 80% cardiac, respectively).83 Some other disease sites such as bone and head/neck had low response rates (20% to 25%). Although the ideal duration of treatment of IFN-α is unknown, up to 2 years of treatment was shown to result in persistent remissions and stable disease in 2 reports.83,84 Considerations that might limit the use of IFN-α include the variability of responses in all disease sites, frequent intolerable side effects in up to 50% of the patients (fatigue, arthralgia, myalgia, depression), and patient aversion to the subcutaneous route of administration. However, IFN remains a viable treatment option, especially when targeted therapies are unavailable.84
Cytokine-directed therapy
Due to the skewing of cytokines and chemokines toward a T-helper-1 (Th1)-pathway in ECD, biologic agents such as interleukin-1 (IL-1) receptor antagonists (anakinra, canakinumab), IL-6 receptor antagonist (tocilizumab), and tumor necrosis factor-α (TNF-α) inhibitors (infliximab, etanercept) have been evaluated as therapeutic agents.85 Of these agents, in 2 case series, anakinra demonstrated a 50% ORR.86,87 However, the responses were variable, with reproducible responses in mild cases of bone, retroperitoneal, and pulmonary ECD but inconsistent in patients with cardiac and CNS/intracranial disease.17,86-88 Similar to IFN-α, subcutaneous injection and intolerable adverse effects in one-third of patients (injection-site rash, headache, nasopharyngitis) may be a barrier to the use of anakinra. The evidence for other biologic agents is quite limited. There have been 2 reported cases of canakinumab, an IL-1β receptor antagonist, in the setting of disease progression despite anakinra; 1 of these 2 had a favorable response.89,90 Infliximab, a TNF-α inhibitor, has been used in several cases with mixed responses. Combining the reported cases, responses were seen in 7 of 23 patients (30%).87,91,92 Another TNF-α antagonist, etanercept, was used in 2 ECD cases without reported response.87 In a small phase 2 clinical trial of the IL-6 antagonist, tocilizumab, 2 of 3 ECD patients had responses in cardiac and retroperitoneal disease whereas 1 patient had progression of CNS disease.93
Cytotoxic chemotherapy
Several chemotherapeutic agents and regimens have been explored in ECD, based on clinical experience in LCH and other hematologic neoplasms. Most of the knowledge and data exist for cladribine, a purine analog, based on several case reports.94-97 In a case series of 21 ECD patients, clinical responses were seen in 52% of patients treated with cladribine with a 9-month median duration of response.98 In this study, there were no specific features predictive of a response to cladribine and responses were seen in all disease sites. Cladribine treatment was well tolerated overall, with a median of 2.5 cycles, guided by the disease response and myelosuppression. Due to the risk of prolonged lymphopenia, we do not recommend >3 to 4 cycles of cladribine. Other less commonly used chemotherapeutics in refractory cases have included cyclophosphamide, vinblastine, high-dose IV methotrexate, lenalidomide, and autologous stem cell rescue with high-dose cytotoxic chemotherapy.99-106
Corticosteroids and immunosuppressants
Corticosteroids are not considered effective as monotherapy for ECD, although they may be used as adjuncts to improve acute symptoms related to tissue swelling such as in the case of orbital disease with impending vision loss. Similarly, weekly oral methotrexate was not shown to be efficacious in a case series of 13 patients, except for prolonged disease improvement and stabilization in 2 patients with ocular (subconjunctival and choroidal) ECD.107 In a French cohort study, treatment with corticosteroids and immunosuppressive agents was not associated with improved survival.82
Surgery and radiation therapy
Due to the multifocal nature of ECD requiring systemic therapies, surgical resection is generally not curative. ECD is not a radiosensitive disease, and we do not recommend this modality either.108 The exceptions to this include situations in which immediate palliation of symptoms is needed (large tumors causing CNS, ocular, or internal organ compromise).
Recommended treatment approach
For patients with multisystem BRAF-V600-mutant ECD with life-threatening cardiac or neurologic involvement, our first-line recommendation is to consider BRAF-inhibitors such as vemurafenib or dabrafenib. The choice of BRAF-inhibitor may be guided by the toxicity profile in light of the particular patient’s clinical status, and the experience of the treating clinician. For BRAF-V600–mutated ECD without end-organ dysfunction, it is appropriate to consider either BRAF-inhibitor or immunosuppressive/cytotoxic therapy, balancing ECD symptom management and side effects of treatment. For patients without BRAF-V600–ECD, we recommend pursuing NGS to evaluate other MAPK-ERK pathway alterations that can be treated with a MEK-inhibitor. “Empiric” treatment with MEK-inhibitors for BRAF-V600–wild-type ECD without an identified MAPK pathway mutation is a reasonable approach for an acutely ill patient with heart/CNS involvement without viable alternative therapies. In patients with CNS involvement, higher doses of targeted (BRAF- or MEK-inhibitor) therapies or dual therapy for BRAF-V600–mutated ECD may be considered to attain robust response and may be tailored subsequently based on tolerance (Table 4). It is to be noted that the outcomes of associated myeloid neoplasms under targeted therapies are not currently known. For patients without access to targeted therapies, IFN-α or PEG–IFN-α present efficacious treatment options, although the latter may be slightly better tolerated. A retrospective cohort review of 165 ECD patients from the French registry reported an overall survival (OS) benefit with the use of IFN-α, PEG–IFN-α, or targeted therapies.7 In patients with mixed histiocytosis (ECD/LCH overlap), however, IFN-α therapy may be suboptimal (given the non-ECD component of disease) and targeted therapies are favored.28 One of the challenges with targeted therapies or IFN treatment is the risk of disease relapse at discontinuation, necessitating a prolonged duration of treatment. Hence, in patients who are clinically fit to receive systemic chemotherapy and/or are unable to access targeted agents or tolerate them, cladribine may be considered as a limited duration treatment to offer sustained response. For patients with low-burden disease involving bones and retroperitoneum, a biologic agent, especially anakinra can be used.
Response assessment and disease surveillance
There are no prospectively validated response criteria for ECD. However, FDG-PET-CT is considered the optimal modality for ECD response assessment.3,16,49 Two recent prospective therapeutic clinical trials in ECD implemented a modified PET Response Criteria in Solid Tumors (PERCIST) and may be used for assessment of response to therapy.3,16 FDG PET imaging should be obtained 3 to 6 months after initiation of therapy to assess metabolic response. Complete metabolic response, that is, normalization of lesion FDG avidity to that of the surrounding organ background, is considered the optimal response to ECD therapy. If achieved, the complete metabolic response may not occur for several months; therefore, continued surveillance FDG-PET may demonstrate ongoing metabolic improvement. The degree of metabolic response varies by patient and treatment regimen. A sustained partial metabolic response in the setting of clinical improvement represents a favorable outcome. Organ-specific imaging such as CT or MRI (eg, heart, brain, orbit) should be performed every 3 to 6 months initially, and every 6 to 12 months once disease stabilizes. Because of the tissue fibrosis associated with ECD, treated lesions may not fully regress, and therefore the degree of anatomic response (ie, shrinkage of lesions by CT/MRI) may not accurately reflect disease activity or response to treatment. This is particularly characteristic of longstanding lesions in the retroperitoneum, abdomen, orbits, and sinuses. Hence, anatomic, metabolic, and clinical responses must be considered in light of one another. C-reactive protein is elevated at diagnosis in 80% of cases,47 and its decline with treatment suggests a favorable response. Inflammatory toxicities of targeted therapies and IFN must be considered in the interpretation of these biomarkers. For dermatologic and endocrine manifestations of ECD, we rely on clinical examination and laboratory tests to assess response. Although skin involvement by ECD may respond to treatments, endocrinopathies are typically permanent. Additionally, monitoring for pituitary hormone abnormalities every 1 to 2 years (Figure 5) is recommended, as endocrinopathies may develop during the course of treatment as well.109 Due to the propensity of ECD to infiltrate the bone marrow and its association with other myeloid neoplasms or clonal hematopoiesis of indeterminate potential,110 clinicians should continually monitor peripheral blood counts and strongly consider bone marrow evaluation with myeloid NGS to assess for abnormalities.21
Apart from the French study previously described,67 which demonstrated that relapse is likely in BRAF-V600–mutant ECD in the setting of complete cessation of vemurafenib without achieving sustained CR, there are currently no data to inform strategies for optimal dosing and duration of treatment of either MEK-inhibitor therapy or conventional therapies. Some ECD clinicians have adopted an intuitive approach of chronic administration of low doses of BRAF- or MEK-inhibitors to maintain a patient’s optimal response with minimal toxicity. Others have attempted intermittent administration of higher doses of targeted therapy in efforts to ameliorate toxicity but maintain adequate treatment. Sequential treatments with targeted therapies followed by conventional therapies have not yet been evaluated but could be an option to prevent mid- or long-term relapses.
Prognosis
The prognosis of ECD can vary based on the site of disease and the response to therapy. In a large French cohort of 165 ECD patients, factors associated with worse OS included advanced age and disease involving the CNS, lungs, and retroperitoneum.7 Advances in diagnosis and therapeutics of ECD are underscored by the tremendous improvement in 5-year OS rates over the last 2 decades, from 43% in a study from 1996 to 83% in a recent study.7,50 With the advent of targeted therapies, even severe manifestations of ECD have become a chronic, rather than fatal, illness. It should be noted that many patients endure persistent symptoms and disabilities even in the setting of complete radiologic responses, and referral for rehabilitation and supportive care can be of benefit. On rare occasions, patients still succumb to advanced disease with irreversible organ damage, especially cerebellar dysfunction.
Conclusion
ECD is a rare histiocytic neoplasm with heterogeneous features, posing significant diagnostic and treatment challenges to clinicians. Multidisciplinary collaboration is often needed for appropriate management; a network of ECD care centers offering specialized care has been created by the Erdheim-Chester Disease Global Alliance (ECDGA; http://erdheim-chester.org/). ECD diagnosis presents unique challenges in its dependence upon interpretation of diverse clinical, pathological, and radiographic characteristics. Lesional tissue should be biopsied in all cases to confirm ECD diagnosis and establish mutational status to potentially guide therapy. Most ECD patients require treatment, and the choice of therapy must be individualized based on clinical characteristics, disease mutational status, and tolerance of treatment. Despite several advances in the understanding of the biology and treatment of ECD, questions about optimal dosing, duration of treatment, and easily accessible biomarkers for assessing treatment response remain unknown. Collaborative work among ECD clinicians and scientists may shed light on these questions in the future.
Acknowledgments
This manuscript is dedicated to Kathy Brewer, founder of the Erdheim-Chester Disease Global Alliance (http://erdheim-chester.org), and ECD patients worldwide as they were the authors’ major inspiration for this work. The authors want to acknowledge the services provided by the Division of Biomedical and Scientific Visualization, Mayo Clinic, for the creation of the illustration used in Figure 5 in our manuscript.
Authorship
Contribution: All authors participated in outlining the manuscript and providing expert recommendation grading; G.G. wrote the manuscript draft with substantial input and supervision from J.H. and E.L.D; all authors then contributed to the editing of the manuscript; and E.L.D. and G.G. provided the radiographic images.
Conflict-of-interest disclosure: M.C. has received honoraria from Mallinckrodt. K.T. received lecture fees from Eisai Co. Ltd., Kyowa Hakko Kirin Co. Ltd., Bristol-Myers Squibb, Celgene K.K., Daiichi Sankyo Co., Nippon Shinyaku Co. Ltd., Chugai Pharmaceutical Company, Ono Pharmaceutical Co. Ltd., Otsuka Pharmaceutical Co. Ltd., and Takeda Pharmaceutical Co. Ltd. F.C.-A. is the PI of a French national academic trial on the efficacy of cobimetinib for wild-type histiocytoses (NCT04007848). A.M.G. receives speaking fees from Seattle Genetics and consulting fees from Jazz Pharmaceuticals, Daiichi Sankyo, Kyowa Kirin, and Navican. M.K. has received consultancy fees from Shionogi & Co. Ltd., Merck Sharp & Dohme K.K. (MSD K.K.), and Chugai Pharmaceutical Co. Ltd.; has received research funding from Pfizer Japan Inc., Otsuka Pharmaceutical Co. Ltd., Chugai Pharmaceutical Co. Ltd., Astellas Pharma Inc., Kyowa Hakko Kirin Co. Ltd., Takeda Pharmaceutical Company Limited, MSD K.K., Teijin Limited, Eisai Co. Ltd., Sumitomo Dainippon Pharma Co. Ltd., Novartis Pharma K.K., Nippon Shinyaku Co. Ltd., Ono Pharmaceutical Co. Ltd., and Bristol-Myers Squibb K.K.; received honoraria directly received from an entity (Shionogi & Co. Ltd.); gave paid expert testimony for Kyowa Hakko Kirin Co. Ltd., Celgene K.K., Bioverativ Japan Ltd., and Daiichi Sankyo Co. Ltd., and held membership on an entity’s board of directors, speaker’s bureau, or advisory committee for MSD K.K., Astellas Pharma Inc., Eisai Co. Ltd., Otsuka Pharmaceutical Co. Ltd., Ono Pharmaceutical Co. Ltd., Yakult Honsha Co. Ltd., Shire Japan K.K., Celgene K.K., Daiichi Sankyo Co. Ltd., Sumitomo Dainippon Pharma Co. Ltd., Takeda Pharmaceutical Co. Ltd., Chugai Pharmaceutical Co. Ltd., Nippon Boehringer Ingelheim Co. Ltd., Bristol-Myers Squibb K.K., Janssen Pharmaceutical K.K., Kyowa Hakko Kirin Co. Ltd., and Nippon Shinyaku Co. Ltd. J.H. is the coprincipal investigator of a French national academic trial on the efficacy of cobimetnib for wild-type histiocytoses (NCT04007848), and discloses unpaid support from Third Rock Ventures outside the submitted work. E.L.D. discloses unpaid support from Third Rock Ventures, outside the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Eli L. Diamond, Department of Neurology, Memorial Sloan Kettering Cancer Center, 160 East 53rd St, 2nd Floor Neurology, New York, NY 10022; e-mail: diamone1@mskcc.org; and Julien Haroche, Department of Internal Medicine 2, Assistance Publique–Hôpitaux de Paris, Pitié-Salpêtrière Hospital, Paris, France; e-mail: julien.haroche@aphp.fr.
