

Key Points
Up to 30% of RUNX1 mutations in the Leucegene AML cohort were confirmed to be germline.
RUNX1 germline–mutated AML shows a high frequency of NRAS mutations and other mutations known to activate various signaling pathways.

Abstract
RUNX1 is mutated in ∼10% of adult acute myeloid leukemia (AML). Although most RUNX1 mutations in this disease are believed to be acquired, they can also be germline. Indeed, germline RUNX1 mutations result in the well-described autosomal-dominant familial platelet disorder with predisposition to hematologic malignancies (RUNX1-FPD, FPD/AML, FPDMM); ∼44% of affected individuals progress to AML or myelodysplastic syndromes. Using the Leucegene RUNX1 AML patient group, we sought to investigate the proportion of germline vs acquired RUNX1 mutations in this cohort. Our results showed that 30% of RUNX1 mutations in our AML cohort are germline. Molecular profiling revealed higher frequencies of NRAS mutations and other mutations known to activate various signaling pathways in these patients with RUNX1 germline–mutated AML. Moreover, 2 patients (mother and son) had co-occurrence of RUNX1 and CEBPA germline mutations, with variable AML disease onset at 59 and 27 years, respectively. Together, these data suggest a higher than anticipated frequency of germline RUNX1 mutations in the Leucegene cohort and further highlight the importance of testing for RUNX1 mutations in instances in which allogeneic stem cell transplantation using a related donor is envisioned.
Introduction
Many acute leukemia predisposition syndromes have been identified.1 The revised World Health Organization classification categorizes myeloid neoplasms with germline predisposition as a distinct entity.2 The most frequently mutated genes in these syndromes are GATA2, ETV6, CEBPA, and RUNX1.3 Inherited mutations in RUNX1 lead to familial platelet disorder with predisposition to hematologic malignancies or RUNX1-FPD. In this disease, alterations in RUNX1 include heterozygous missense, frameshift, and non-sense mutations, as well as large intragenic and chromosomal deletions involving chr21q22.12.4,5 It is estimated that ∼44% of individuals with RUNX1-FPD will develop acute myeloid leukemia (AML) or myelodysplastic syndromes, with a median age of onset of 33 years.6,7 Identifying these patients is crucial for genetic counseling and for carefully monitoring patients at risk for malignant transformation to optimize the timing of treatment intervention and, if an allogeneic transplantation is indicated, to ensure that an affected family member is not a donor.1,3
Study design
Material and methods are detailed in supplemental Materials and methods (available on the Blood Web site). Primary AML specimens were collected between 2001 and 2015, according to Quebec Leukemia Cell Bank (BCLQ) procedures. Genetic variants were identified by RNA-sequencing, as described,8 and validated in tumor DNA. Copy number variations (CNVs) were identified using whole-genome sequencing (depth coverage ∼5×). Forty-four RUNX1 mutations and 12 non-RUNX1 mutations were validated as somatic or germline based on polymerase chain reaction and bidirectional Sanger sequencing of patient normal DNA obtained from buccal swabs or saliva harvested at diagnosis (oligo sets are described in supplemental Tables 1 and 2). Differential gene expression analysis and screening of acquired mutations in 80 AML-related genes were performed and used to compare groups of patients carrying germline and somatic RUNX1 mutations.
Results and discussion
High frequency of RUNX1 germline mutations in adult AML
The algorithm for somatic (newly acquired) vs germline status of RUNX1 mutations in our 430 specimens from the Leucegene collection is shown in Figure 1A. In brief, 67 specimens were found with RUNX1 mutations. Of these, 23 were excluded for 3 main reasons: (1) the identified RUNX1-mutated allele was considered polymorphic in the general population, (>1E−2; n = 19; supplemental Table 3); (2) there was insufficient specimen material for confirmation studies (n = 2), and (3) variant allele frequency (VAF) was <30% (n = 2) (Figure 1A; supplemental Table 4). Next, comparison of RUNX1 allele status was performed for each of the 44 remaining specimens using normal and tumor DNA (Figure 1A). Chromatogram examination was performed and analyzed individually to identify germline candidate specimens, as detailed in Figure 1B and supplemental Figures 1 and 2.
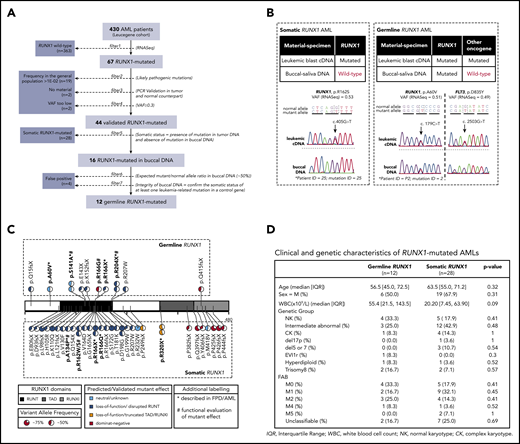
AML patients from Leucegene cohort carrying germline and somatic RUNX1 mutations. (A) Pipeline used for the identification of germline and somatic RUNX1 mutations. (B) Sequencing chromatograms of leukemic complementary DNA (cDNA)/normal DNA pairs covering mutation sites for RUNX1 and control oncogene. Refer to supplemental Figures 1 and 2 for a complete analysis of leukemic/normal DNA pairs. (C) Primary structure of RUNX1 protein with identified germline mutations (upper panel) and somatic mutations (lower panel). Pie charts show VAF for each mutation, as revealed by RNA-sequencing. Predicted effect of mutation on protein function was determined by prediction tools when not previously described in functional studies and it is depicted by the color scheme. Mutations that have been validated in functional studies are highlighted in bold type and are marked with a pound sign (#); mutations that have been described in RUNX1-FPD pedigrees are highlighted in bold type and are marked with an asterisk (*). Refer to supplemental Table 6 for the references to such studies. Protein domains and mutation positions are based on isoform NP_001745.2. RUNT: 85-206, TAD: 318-398, RUNXI: 389-480. (D) Clinical and genetic characteristics of AML patients with germline and somatic RUNX1 mutations. C, complex karyotype; FAB, French-American-British; IQR, interquartile range; M, male; NK, normal karyotype; PCR, polymerase chain reaction; WBC, white blood cell count.
AML patients from Leucegene cohort carrying germline and somatic RUNX1 mutations. (A) Pipeline used for the identification of germline and somatic RUNX1 mutations. (B) Sequencing chromatograms of leukemic complementary DNA (cDNA)/normal DNA pairs covering mutation sites for RUNX1 and control oncogene. Refer to supplemental Figures 1 and 2 for a complete analysis of leukemic/normal DNA pairs. (C) Primary structure of RUNX1 protein with identified germline mutations (upper panel) and somatic mutations (lower panel). Pie charts show VAF for each mutation, as revealed by RNA-sequencing. Predicted effect of mutation on protein function was determined by prediction tools when not previously described in functional studies and it is depicted by the color scheme. Mutations that have been validated in functional studies are highlighted in bold type and are marked with a pound sign (#); mutations that have been described in RUNX1-FPD pedigrees are highlighted in bold type and are marked with an asterisk (*). Refer to supplemental Table 6 for the references to such studies. Protein domains and mutation positions are based on isoform NP_001745.2. RUNT: 85-206, TAD: 318-398, RUNXI: 389-480. (D) Clinical and genetic characteristics of AML patients with germline and somatic RUNX1 mutations. C, complex karyotype; FAB, French-American-British; IQR, interquartile range; M, male; NK, normal karyotype; PCR, polymerase chain reaction; WBC, white blood cell count.
To rule out leukemia contamination of nonleukemic tissue as the likely explanation for false-positive results, we evaluated normal DNA/AML complementary DNA pairs, from each patient, for the presence of an additional genetic marker (eg, NRAS, FLT3) that, for germline RUNX1 mutations, should be detectable in the leukemic DNA and not in the normal counterpart (Figure 1B). Additionally, heterozygous germline mutations should show the expected mutant/normal allele ratio in buccal DNA of near 50% VAF. Four specimens were considered false positives and were excluded from analysis. To control RUNX1 copy number status, CNVs and ploidy were determined using low-pass whole-genome sequencing data (supplemental Materials and methods; supplemental Figure 3) and/or cytogenetics information (supplemental Table 5). With this approach we could confidently validate 12 candidate specimens as positive for the presence of germline RUNX1 mutations (Figure 1A; supplemental Figure 2). This shows, at least in our cohort, that up to 30% of RUNX1 mutations are germline. If our VAF filter is not taken into consideration (as per certain clinical laboratories), this frequency would be 28.6% (12/42).
RUNX1 germline mutation characteristics
Germline RUNX1 mutations included missense (n = 5/12), non-sense (n = 4/12), and frameshift (n = 3/12) mutations (Figure 1C). Among these, only 4 germline RUNX1 variants have been previously reported in RUNX1-FPD (p.A60V, p.S141A, p.R166X, and p.R204X).9-12 Of the 10 distinct germline RUNX1 mutations identified in this study, 8 are predicted to be deleterious to RUNX1 function (based on published functional studies or prediction tools), and 6 are predicted to be pathogenic/likely pathogenic based on MM-VCEP rules13 (supplemental Tables 6 and 7). Functional predictions for RUNX1 mutations are in agreement with abnormal expression levels of several transcripts associated with the RUNX1mut signature (supplemental Figure 4).
Clinical and molecular characteristics of germline and somatic RUNX1–mutated patients
Germline RUNX1-mutated patients showed a tendency to develop leukemia at a younger age (median, 56.5 years) than somatic RUNX1 AML patients (median, 63.5 years) and to have higher white blood cell counts (Figure 1D).14-16 Most notably, 4 of 12 patients (33.3%) in the germline RUNX1 group developed leukemia at younger than 50 years of age, compared with only 2 patients from the somatic RUNX1 group (7.1%; supplemental Figure 5A). No major difference in French-American-British or genetic subtype frequency was observed between the 2 groups. Two germline mutations, p.S141A and p.R204X, were recurrent in >1 patient. Patient data confirmed that patients P3 and P4 carrying p.S141A were related (see “Germline mutations in GATA2 and CEPBA observed in AML patients with early onset”). On the other hand, family history for patients carrying p.R204X (P9 and P10) was not available, and pairwise comparison of genetic alterations in common between each specimen of the Leucegene cohort could not differentiate them from random unrelated pairs (supplemental Figure 6).
Somatic mutations in RUNX1 are frequently observed in leukemic progression of individuals with germline RUNX1 mutation.17 This was indeed found in 2 of our patients (Figure 2A; supplemental Figure 5A). We observed a trend for higher frequency of acquired NRAS mutations in germline RUNX1 specimens (P = .055) than in somatic RUNX1 specimens. Mutations in NRAS have been observed in malignant transformation of RUNX1-FPD patients.18 Interestingly, we observed a higher frequency of activating mutations, notably in the RAS pathway, for the germline subgroup (41.7%) compared with the somatic subgroup (21.4%; P = .254) (Figure 2B-C). ASXL1 was mutated more frequently in somatic RUNX1–mutated specimens (39.3% vs 25% in the germline group; P = .484). Another group identified that co-occurring mutations in RUNX1-mutated AML patients were primarily ASXL1 mutations in older patients and RAS mutations in younger patients.19 This dichotomy in RUNX1 co-occurring mutations indicates that several mechanisms of malignant progression may be at play for different groups of RUNX1-mutated patients.

Mutational profile of primary AML cells with germline RUNX1mut shows enrichment of activated signaling pathway. (A) Mutation grid of RUNX1-mutated primary AML specimens showing 1 patient sample per column. Samples are grouped according to their RUNX1 mutation germinal status. Co-occurring mutated genes are shown in each row and are grouped by gene ontology. Enrichment between comparison groups was calculated using the Fisher’s exact test. Distribution of the most frequent mutated genes (B) and mutated groups (C) in germline and somatic RUNX1 patient cohorts. TF, transcription factor.
Mutational profile of primary AML cells with germline RUNX1mut shows enrichment of activated signaling pathway. (A) Mutation grid of RUNX1-mutated primary AML specimens showing 1 patient sample per column. Samples are grouped according to their RUNX1 mutation germinal status. Co-occurring mutated genes are shown in each row and are grouped by gene ontology. Enrichment between comparison groups was calculated using the Fisher’s exact test. Distribution of the most frequent mutated genes (B) and mutated groups (C) in germline and somatic RUNX1 patient cohorts. TF, transcription factor.
Differential gene expression analysis identified 10 genes that were significantly upregulated in the germline subgroup (false discovery rate < 0.01; supplemental Figure 7; supplemental Table 8). Interestingly, IL11, which plays important roles in the differentiation and maturation of megakaryocytes, was among the top candidates overexpressed in the germline RUNX1 group compared with the somatic RUNX1 group.
Germline mutations in GATA2 and CEPBA observed in AML patients with early onset
Remarkably, the youngest patient to develop leukemia in our cohort at age 27 years (P3) carried a germline CEBPA mutation (p.R297L). The p.R297L mutation has been previously reported as germline in familial AML with mutated CEBPA.20 Of interest, his mother carrying the same germline mutation load developed AML at 59 years of age and acquired a GATA2 mutation not found in the son’s specimen (supplemental Figure 5B,D; supplemental Table 9). This suggests that RUNX1 and CEBPA are not sufficient to induce full transformation of hematopoietic stem/progenitor cells and that additional events, such as GATA2, are involved. Conversely and interestingly, a patient with germline GATA2 mutation developed AML at age 31 years (P42) in which acquired RUNX1 mutation was found (supplemental Figure 5A,C).
Using rigorous criteria, we could identify a high incidence of 30% germline RUNX1 mutations in RUNX1-mutated AML of the Leucegene cohort. RNA-sequencing at the time of AML diagnosis revealed that these leukemias show a high frequency of NRAS mutations and of other mutations known to activate various signaling pathways. Identification of germline mutations in other driver genes, such as CEBPA and GATA2, further highlight the notion that germline mutations might be underestimated and, importantly, impact leukemia predisposition. Overall, and at least this study suggests that testing for donor RUNX1 mutation status may be important in families in which RUNX1-FPD cases have been identified and familial stem cell transplantation is considered. These data merit confirmation in larger population cohorts.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Katrin Ericson for helpful discussions and critical reading of the manuscript, Muriel Draoui for project coordination, and Sophie Corneau and Nadine Mayotte for sample coordination. The authors acknowledge Raphaëlle Lambert and Jennifer Huber at the Institute for Research in Immunology and Cancer genomics platform for RNA sequencing, as well as the BCLQ team who characterized and provided all AML samples from the Leucegene cohort, with special thanks to G. D’Angelo, C. Rondeau, and S. Lavallée.
This work was supported by the Leukemia and Lymphoma Society and the Babich Family Foundation Runx1 Research Program (grant 6553-18), the Government of Canada through Genome Canada, and the Ministère de l’Économie et de l’Innovation du Québec through Génome Québec (grant 13528). A research grant from the Canadian Cancer Society (grant 705476) also funded part of this project in collaboration with The Cole Foundation, the Molson Foundation, the R. Howard Webster Foundation, the Mirella & Lino Saputo Foundation, the Birks Family Foundation, the Letko Brosseau’WCPD Foundation, the Université de Montréal, the Trottier Foundation, the Maryse & William Brock Chair, the Power Corp, the Fondation CHU Sainte Justine, the Montreal Children’s Hospital Foundation, the Morris & Rosalind Goodman Family Foundation, the Zeller Family Foundation, the Drummond Foundation, the David Laidley Foundation, and the Henry & Berenice Kaufmann Foundation. J.H. is the recipient of the Industrielle-Alliance research chair in leukemia at Université de Montréal. BCLQ is supported by grants from the Cancer Research Network of the Fonds de Recherche du Québec–Santé. L.S. received graduate scholarships from the Cole Foundation and the Fonds de Recherche du Québec–Santé. V.-P.L. is the recipient of a Vanier Canada Graduate Scholarship. J.-F.S. is supported by a postdoctoral fellowship from the Canadian Institutes of Health Research (grant 158159).
Authorship
Contribution: L.S., J.-F.S., J.H., and G.S. designed the experiments and wrote the manuscript; L.S., V.-P.L., and I.B. performed validation of mutations; L.S., J.-F.S., and C.-Y.Y. performed mutation, CNV, and transcriptomic characterization and generated all of the figures; G.S. and M.-E.B. spearheaded the data analysis and manuscript redaction; and V.-P.L., G.B., E.A., and S.L. assisted with data analysis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Guy Sauvageau, PO Box 6128, Station Centre-Ville, University of Montreal, Montreal H3C3J7, QC, Canada; e-mail: guy.sauvageau@umontreal.ca; and Josée Hébert, Quebec Leukemia Cell Bank, Research Centre, Maisonneuve-Rosemont Hospital, 5415 L’Assomption Blvd, Montreal, QC, Canada, H1T 2M4; e-mail: josee.hebert@umontreal.ca.
