

In this issue of Blood, evaluated 430 samples from patients with acute myeloid leukemia (AML) for germline and somatic mutations in RUNX family transcription factor 1 (RUNX1). They found that nearly 30% of the identified variants were germline.1
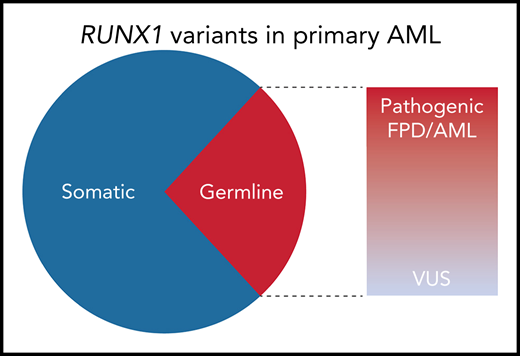
Among the nonpolymorphic RUNX1 variants identified in 430 adult AML specimens from the Leucegene AML cohort, 29% were germline. Of the 10 distinct germline variants identified, 6 were classified as pathogenic or likely pathogenic by the MM VCEP criteria; 8 of the 10 were predicted to be deleterious to RUNX1 function by functional studies or prediction algorithms. VUS, variant of unknown significance.
Among the nonpolymorphic RUNX1 variants identified in 430 adult AML specimens from the Leucegene AML cohort, 29% were germline. Of the 10 distinct germline variants identified, 6 were classified as pathogenic or likely pathogenic by the MM VCEP criteria; 8 of the 10 were predicted to be deleterious to RUNX1 function by functional studies or prediction algorithms. VUS, variant of unknown significance.
The importance and the unique biology of RUNX1 in AML have been recognized by the World Health Organization (WHO) by adding 2 separate categories of AML with RUNX1 mutations in the 2016 WHO classification of hematologic neoplasms.2 AML with mutated RUNX1 makes up ∼10% of newly diagnosed AML and is associated with poor event-free and overall survival.3 Although the vast majority of RUNX1 mutations are presumed to be somatic, germline mutations in RUNX1 cause a rare leukemia predisposition syndrome called familial platelet disorder (FPD) with associated myeloid malignancy (FPDMM, also referred to as FPD/AML), acknowledged in its own WHO category.
Despite the growing awareness of leukemia predisposition syndromes, their prevalence in patients diagnosed with de novo AML or myelodysplastic syndrome (MDS) remains poorly defined. Published reports estimate that 4% to 9% of adults with myeloid neoplasms have germline predisposition.4,5 More recent analyses of larger cohorts of patients with myeloid malignancies suggest that germline myeloid malignancy predisposition syndromes may be even more common.6-8 Until now, the prevalence of germline RUNX1 mutations in AML was largely unexplored.
To determine the frequency of RUNX1 variants in primary AML, Simon et al sequenced 430 adult leukemia samples from the Quebec Leukemia Cell Bank (Leucegene cohort). After excluding common polymorphisms, 48 patients (11% of the Leucegene cohort) had RUNX1 variants that were either absent from or exceedingly rare in the general population. To determine which of these were present in the germline, the authors sequenced the mutated regions in paired buccal DNA. An elaborate genotyping scheme was used to exclude false positives resulting from hematopoietic cell contamination, with non-RUNX1 somatic mutations serving as a marker of contaminating leukemic DNA. By using this approach, 12 of 42 evaluable RUNX1 variants (∼29%) were confirmed to be germline (see figure). Among the 40 RUNX1 variants present at a high (≥0.3) allelic fraction, 30% were germline.
The strikingly high frequency of germline RUNX1 variants in the Leucegene cohort deserves attention. Perhaps the closest comparison is a study from the University of Chicago, in which the investigators assessed the germline status of variants in familial leukemia–associated genes identified by a "somatic" next-generation sequencing (NGS) panel in 360 patients with hematologic malignancies.9 By applying the filtering criteria from Simon et al, the AML patients in the Chicago cohort had 20 nonpolymorphic RUNX1 variants, of which only 2 (10%) were germline. If restricted to mutations with an allelic fraction ≥0.3, germline variants accounted for 14% (2 of 14) of variants. The 2 cohorts were different enough to preclude direct comparison. Nonetheless, the observed differences in RUNX1 variant frequencies between the 2 studies are not statistically robust at this relatively small sample size, which underscores the need for future validation with more patients who have RUNX1-mutated AML.
Because of the high allelic heterogeneity of germline RUNX1 variants, many of which have been reported only in single patients or families, the determination of variant pathogenicity requires caution.3 A personal and family history of life-long thrombocytopenia, platelet dysfunction, and an autosomal dominant pattern of familial AML can go a long way in supporting a diagnosis of FPD/AML; however, this information is not available for members of the Leucegene cohort. This quandary is not uncommon in the clinic when evaluating a patient with RUNX1-mutatedAML, where a germline mutation could have arisen de novo, or family history could be unknown or incomplete. The recently published ClinGen Myeloid Malignancy Variant Curation Expert Panel (CG MM-VCEP) recommendations for interpreting the pathogenicity of RUNX1 germline variants put forth a set of rules to systematically evaluate a RUNX1 variant.10 Applying the CG MM-VCEP curation rules, 6 of 10 distinct germline variants identified by Simon et al were predicted to be pathogenic or likely pathogenic, and 8 of 10 were predicted to be deleterious to RUNX1 function by functional studies or prediction algorithms. The nuance in interpreting RUNX1 variant pathogenicity is illustrated by a family in the Leucegene cohort, in which 2 first-degree relatives developed AML and were found to carry germline variants in both RUNX1 and CEBPA genes, previously reported in their respective familial leukemia syndromes. Both patients also acquired second-hit somatic CEBPA mutations, making a case that in this family, the germline RUNX1 variant may serve as a modifier for primarily CEBPA-driven leukemia.
Where does this leave us with respect to germline RUNX1 mutations or—even more broadly—germline mutations in familial leukemia-associated genes in 2020? With great testing power comes great responsibility. Identifying germline predisposition can significantly impact patient care in positive ways, starting with a more accurate interpretation of bone marrow morphology and prognostic significance of somatic alterations within the context of a known familial syndrome. Awareness of syndrome-specific hematopoietic and extra-hematopoietic complications, such as platelet dysfunction in RUNX1 mutation carriers or predisposition to liver cirrhosis in telomere biology disorders, will impact management and disease surveillance. Finally, accurate genetic diagnosis is important for treatment decisions, including donor choice, and, in certain situations, the timing and choice of a conditioning regimen for allogeneic transplantation.
To accomplish all of this, we have to get better at performing and interpreting germline variant testing. The gold standard of cultured skin fibroblasts takes several weeks and requires a facility to culture skin fibroblasts. If not started soon after AML diagnosis, testing may lead to a costly delay at the time of transplant evaluation for an AML patient. As we enter the golden age of AML predisposition genetics, we need improvement in 3 major areas. To expand, simplify, and speed up genetic evaluation, "somatic" NGS panels could be expanded to enhance the capture of genes linked to MDS/AML germline predisposition syndromes. When nonpolymorphic variants in RUNX1 or other predisposition-associated genes are identified in a blood or marrow specimen, we need to have protocols to ensure appropriate reflexing to confirmatory germline testing. Finally, a centralized, expert-run variant interpretation service could go a long way toward expanding access to evidence-based variant interpretation for rare and emerging MDS/AML predisposition syndromes.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
