

In this issue of Blood, report the largest ever cohort series using eculizumab, a humanized monoclonal immunoglobulin G antibody inhibitor of complement protein C5, as first-line therapy for pediatric patients with high-risk transplant-associated thrombotic microangiopathy (TA-TMA).1 These researchers were innovators in TA-TMA research by originally defining biologically linked, biomarker-driven diagnostic criteria and using these criteria to routinely monitor for TA-TMA and to inform practice-changing treatment strategies.2
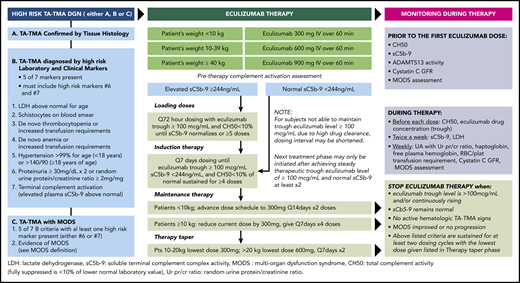
Comprehensive standard-of-care diagnostic, therapeutic, and monitoring program used to treat pediatric patients with TA-TMA. See Figure 1 in the article by Jodele et al that begins on page 1049.
Comprehensive standard-of-care diagnostic, therapeutic, and monitoring program used to treat pediatric patients with TA-TMA. See Figure 1 in the article by Jodele et al that begins on page 1049.
TA-TMA is a representative example of a toxicity syndrome linked to overactivation of complement pathways and endothelial injury resulting in thrombosis, hemolysis, and often multiorgan dysfunction syndrome that has historically been associated with considerable morbidity, high rates of health care use, and mortality after allogeneic hematopoietic cell transplantation (allo-HCT).2,3 TA-TMA has been difficult to reliably diagnose and treat in clinical practice, given an initially limited understanding of its pathobiology; variable, nonspecific, and inconsistently adopted diagnostic criteria; and for quite some time, ineffective therapies.4
By using their previously defined criteria, the authors identified 177 (31%) of 566 patients who developed TA-TMA after allo-HCT between 2012 and 2018 at their center. Of these patients, 64 (36%) were deemed to have high-risk TA-TMA which, on the basis of their previous work, would be expected to result in a dismal 1-year overall survival of <20%.2 With an innovative pharmacokinetically (PK) and pharmacodynamically (PD)–targeted therapeutic strategy, 41 patients (64%) with high-risk TA-TMA responded to eculizumab, with the majority (56%) achieving complete remission. Patients with the highest levels of complement activation, as measured by elevated serum sC5b-9, at TA-TMA diagnosis and at the start of therapy were less likely to respond to and required longer courses of eculizumab. Even in eculizumab responders, the authors detail the substantially increased toxicities faced by these patients, including fluid overload with serositis, gastrointestinal bleeding, central nervous system symptoms, acute kidney injury requiring renal replacement therapy, and the need for frequent anti-hypertensives and blood product transfusions. About one-third of treated patients developed a bacterial bloodstream infection, a rate comparable to that of patients with untreated TA-TMA. Although fungal infections were seen in 6 patients, most had concurrent steroid-refractory graft-versus-host disease (GVHD), which required additional immunosuppression, suggesting that eculizumab therapy may not greatly increase the risk of serious infections. In the most remarkable result of the study, overall survival at 1 and 3 years was 66% and 53%, respectively, a stark improvement when compared with that in patients with untreated TA-TMA in the era before complement inhibition.2
What makes this study so compelling is the strong evidence that a rigorous prospective TA-TMA diagnostic, monitoring, and treatment program targeting high-risk patients can turn a previously nearly universally fatal condition into a more manageable one for several reasons (see figure). First, it allows for more frequent and earlier identification of TA-TMA post-HCT and more readily stratifies patients into low- and high-risk groups for further study. Second, the PK/PD-targeted approach allows for more precise, personalized dosing that is driven by both clinical and serum biomarker responses. This manner of dosing may have the added benefit of limiting the potentially unnecessary and expensive health care costs of continual administration of eculizumab. Third, this strategy demonstrates that effective therapy is not one sizes fits all, and instead mirrors the biology of TA-TMA, which represents a spectrum of severity and clinical manifestations.5 Moreover, low-risk TA-TMA, although not a focus of the study, may result in end-organ damage such as chronic renal injury, which has known independent associations with higher mortality after allo-HCT.6 It would be of interest for the authors to report the outcomes of those with low-risk TA-TMA, because these patients may also incur considerable toxicities and have inferior outcomes when compared with patients who never develop TA-TMA.
Despite these data, the accurate incidence of TA-TMA after allo-HCT remains incompletely understood. About 72% of patients who developed TA-TMA in the study underwent allo-HCT for nonmalignant hematologic conditions, and importantly, all were pediatric patients. In contrast, the majority of allo-HCTs performed are for adults with malignant hematologic conditions, which may partly explain why a TA-TMA incidence of 31% seems higher than expected.2 The clear link between GVHD and TA-TMA reported in the Jodele et al study and many others illustrates that development of TA-TMA is likely dependent on numerous factors, including a patient’s inherent genetic susceptibility (complement gene mutations), the type of allo-HCT performed, the GVHD prophylaxis used, and the incidence of acute GVHD inherent to the specific HCT platform chosen.7,8 Therefore, only large prospective trials or querying large registry data sets with heterogeneous patient and transplantation characteristics will allow us to determine the truest incidence of TA-TMA across a broader population and to more clearly define the risk factors associated with TA-TMA in multivariable analyses.
Many questions remain. For example, why do roughly one third of high-risk patients not respond to eculizumab therapy? Perhaps there was suboptimal inhibition of C5, or perhaps these patients began therapy too late in their TA-TMA disease course to derive benefit, as suggested by the authors. Maybe the clues lie in other areas of the complement activation cascade that are more specifically targeted by 1 of the several novel complement pathway inhibitors currently in clinical development.9 Conceivably, because complement disorders often require a second hit in a predisposed patient, perhaps eculizumab nonresponders had ongoing uncontrolled TA-TMA triggers, such as steroid-refractory GVHD, that perpetuated complement activation and endothelial injury.10 Although the HCT field continues to investigate these questions and strives toward a more complete understanding of TA-TMA, the elegant and well-designed treatment roadmap from Jodele et al should be considered a gold standard. Whether their comprehensive approach is reproducible in an adult population and across allo-HCT centers remains to be seen. At the very least, these data should inspire experts across the HCT field to develop harmonized, universally adaptable diagnostic consensus criteria through collaborative, multicenter efforts with the goal of designing definitive prospective clinical trials for the prevention and treatment of TA-TMA. For now, we offer our genuine compliments for another major step toward reducing serious complications and nonrelapse mortality after allo-HCT.
Conflict-of-interest disclosure: M.S. served as a consultant for McKinsey & Company, Angiocrine Bioscience, Inc., and Omeros Corporation, received research funds from Angiocrine Bioscience, Inc., and served on an ad hoc advisory board for Kite (a Gilead company). S.G. served as a consultant for Amgen, Actinium, Celgene, Johnson & Johnson, Takeda, Jazz Pharmaceuticals, Novartis, Kite (a Gilead company), and Spectrum Pharmaceuticals and has received research funding from Amgen, Actinium, Celgene, Johnson & Johnson, Takeda, and Miltenyi Biotec.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal