

Key Points
Macrophage migration in inflammation is highly dependent on fibrinolysis.
Fibrinogen deficiency or elimination of the αMβ2-binding motif from fibrin(ogen) rescues macrophage migration in plasminogen-deficient mice.

Abstract
Efficient migration of macrophages to sites of inflammation requires cell surface–bound plasmin(ogen). Here, we investigated the mechanisms underlying the deficits of plasmin(ogen)-mediated macrophage migration in 2 models: murine thioglycollate-induced peritonitis and in vitro macrophage migration. As previously reported, macrophage migration into the peritoneal cavity of mice in response to thioglycollate was significantly impaired in the absence of plasminogen. Fibrin(ogen) deposition was noted in the peritoneal cavity in response to thioglycollate, with a significant increase in fibrin(ogen) in the plasminogen-deficient mice. Interestingly, macrophage migration was restored in plasminogen-deficient mice by simultaneous imposition of fibrinogen deficiency. Consistent with this in vivo finding, chemotactic migration of cultured macrophages through a fibrin matrix did not occur in the absence of plasminogen. The macrophage requirement for plasmin-mediated fibrinolysis, both in vivo and in vitro, was negated by deletion of the major myeloid integrin αMβ2-binding motif on the γ chain of fibrin(ogen). The study identifies a critical role of fibrinolysis in macrophage migration, presumably through the alleviation of migratory constraints imposed by the interaction of leukocytes with fibrin(ogen) through the integrin αMβ2 receptor.
Introduction
Plasmin(ogen) is essential for maintaining tissue homeostasis1-4 and has a critical role in a plethora of tissue repair and remodeling processes.5-13 Plasminogen also modulates multiple disease processes, especially those caused by irregular inflammation such as asthma,14 arthritis,15 Alzheimer disease,16 and multiple sclerosis.17 Plasmin is a promiscuous enzyme with many potential substrates that could promote the inflammatory response, including fibrin, protease-activated receptors (PARs),18,19 complement factor C5,20 and other proteases.21,22 Fibrin, the main physiological target of plasmin, also has notable contributions to the inflammatory response.23-27 Indeed, many of the plasmin(ogen)-mediated modifications of inflammatory responses are mechanistically linked to fibrin.6,15
Plasmin(ogen) has been increasingly linked to macrophage biology,28,29 which is one possible mechanism connecting plasminogen to inflammation. Macrophage migration is distinctly abnormal in mice lacking plasminogen.28 Localization of plasmin(ogen) to the cell surface, via plasmin(ogen) receptors, is required for effective macrophage migration.30-35 Perturbation of these receptors, via blocking antibodies, gene disruption, or suppression of externalization, impairs macrophage migration. Furthermore, plasmin(ogen) promotes macrophage phagocytosis.29
Plasmin has many potential targets that could influence macrophage migration. In the setting of sterile peritonitis, matrix metalloproteinase 9 (MMP9) activation was notably diminished in plasminogen-deficient mice.22 Replenishment of activated MMP9 partially restored macrophage migration in the absence of plasminogen. PAR-1 activation has also been noted to contribute to plasmin-mediated macrophage migration.18 These findings, however, do not preclude other plasmin substrates as relevant for macrophage migration.
Here, we hypothesized that plasmin-mediated fibrinolysis contributes to macrophage migration. To test this hypothesis, we examined both in vivo macrophage migration in sterile peritonitis and in vitro migration through fibrin matrices. We found that impaired fibrinolysis is a key impediment to macrophage migration under plasminogen-deficient conditions.
Methods
Reagents and cells
Details regarding the reagents and cells used are given in the supplemental Methods (available on the Blood Web site).
Mice
Plasminogen-deficient (Plg−/−), fibrinogen-deficient (Fib−/−), Fibrinogenγ390-396A (Fibγ390-396A), and fXIII-deficient (FXIII−/−) mice have been previously described.3,26,36-39 All studies in non-inbred mice were littermate controlled. Mice at Cincinnati Children’s Research Foundation (CCRF) were all in a C57BL/6J background. At the National Institutes of Health (NIH), mice were in a C57BL/6J background, except for Plg–/Fib– mice and their Plg–/Fib+, Plg+/Fib–, and Plg+/Fib+ littermates, which were in the mixed Black-Swiss/C57BL/6J background. Mice were genotyped as previously described.3,26,36-39 All experiments were performed under approved protocols in vivaria certified by the Association for Assessment and Accreditation of Laboratory Animal Care International and were approved by the Institutional Animal Care and Use Committee before initiation of experimentation.
Induction of peritonitis
Sterile peritonitis was induced with thioglycollate as previously described.28 Here, 200 or 500 µL of 4% Brewer thioglycollate medium (BD Difco) was injected intraperitoneally into mice 8 to 12 weeks of age. Mice were euthanized 72 hours later, and the peritoneal cavity was lavaged with 5 mL of phosphate-buffered saline (PBS).
Differential cell counts
Total cell counts were determined, and cytospins were performed and stained with Diff-Quik (Dade Behring). An observer blinded to genotype performed total cell counts and lavage differentials.
Flow cytometry
Peritoneal lavage was stained with the Live/Dead Cell Viability assay (Thermo Fisher Scientific), and cell surface markers were stained with the following anti-mouse antibodies at 1:100 dilution per 106 cells: Ly6C (AL-21; BD Biosciences), Ly6G (1A8; BD Biosciences), CD86 (GL-1; BioLegend), CD11B (M1/70; BioLegend), CD64 (X54-5/7.1; BioLegend), MHCII (M5/11.4.15.2; eBioscience), F4/80 (BM8; eBioscience), CD11C (N418; eBioscience), and CD45 (30.F11; eBioscience). All samples were analyzed by using a FACS Fortessa cytometer (BD Biosciences). Data analysis was performed by using FlowJo software (Tree Star).
Fibrin(ogen) immunofluorescence
Immunofluorescence confocal microscopy was performed by using paraffin-embedded anterior abdominal wall (including the parietal peritoneum, 3 muscle layers and the connective tissue layer) samples with 5-µm sections. Antigen retrieval in 10 mM citric acid buffer was used, and sections were blocked with 2% bovine serum albumin in PBS for 1 hour. Sections were incubated with anti-fibrin(ogen) antibody (1:500)40 for 1 hour at room temperature and washed in PBS (3 times). Secondary antibody (Rhodamine Red-X anti-rabbit; Jackson ImmunoResearch Laboratories, 711-296-152, 1:500) and 4′,6-diamidino-2-phenylindole (1:500) were added for 1 hour at room temperature. Sections were then washed in PBS (3 times), and #1.5 coverslips were mounted with ProLong Gold Antifade Reagent (Thermo Fisher Scientific). Images were captured on an inverted Nikon A1R+ confocal microscope (40×) using NIS-Elements software (Nikon). Average fluorescent intensity was measured by using NIS-Elements software. A fixed-area square region of interest was drawn around the tissue, and the time measurement analysis tool was used to measure average fluorescent intensity within the region of interest for the AI555 channel. Five individual fields were measured per tissue, and mean fluorescent intensity values were calculated in Microsoft Excel for Mac (version 16.15).
Immunohistochemistry
Details regarding immunohistochemistry are given in the supplemental Methods.
Determination of fibrin(ogen) levels
Peritoneal lavage supernatant fluid was assayed for fibrinogen levels via enzyme-linked immunosorbent assay (Innovative Research). Immunoblotting of fibrin(ogen) was performed with a rabbit anti-fibrinogen antibody (that recognizes both fibrin and fibrinogen) as previously described.39
Macrophage transwell migration
RAW 264.7 cells and bone marrow–derived macrophages (BMDMs) were maintained in culture with Dulbecco’s modified Eagle medium or RPMI 1640, respectively, supplemented with 10% fetal bovine serum. BMDM media were also supplemented with 50 µg/mL macrophage colony-stimulating factor. Reagents were suspended in macrophage migration buffer (MMB) for each experiment. The MMB consisted of Dulbecco’s modified Eagle medium/RPMI supplemented with 0.2% fetal bovine serum. Before the transwell migration assay, 150 µL of a solution of fibrinogen 500 µg/mL, factor XIIIa 5 µg/mL, and batroxobin 20 batroxobin units/mL in MMB was allowed to polymerize in the upper chamber of the transwell chamber (5 µm pores) overnight at 37°C. Cells (2 × 105) were suspended in MMB and placed in the upper chamber, with or without 0.1 µM plasminogen. MMB with 50 ng/mL C5a with 0.1% bovine serum albumin as a carrier was placed in the lower chamber. The transwell system was incubated overnight at 37°C, and the membranes were removed and stained with Diff-Quik. Photomicrographs were captured on a Nikon microscope equipped with NIS-Elements software. Transwell membrane images were generated by using automated stitching of multiple 10× images using the NIS-Elements software. Quantitation of imaging was performed by using the Fiji configuration of ImageJ (NIH).
Statistical analysis
For all except the in vitro assays, the one-way analysis of variance, unpaired 2-tailed statistical analysis was performed on Prism (GraphPad Software). For in vitro assays, 2-tailed, Mann-Whitney U tests were performed on Prism.
Results
Plasminogen deficiency leads to reduced monocyte/macrophage and dendritic cell recruitment to the peritoneal cavity
Migration of leukocytes (primarily monocytes/macrophages) in response to inflammation is diminished in Plg−/− animals.14,22,28 We first sought to characterize this diminished migration in a model of sterile, thioglycollate-induced peritonitis. Forward-scatter (FSC) vs side-scatter (SSC) plots depict distribution of all cell populations 72 hours after induction of thioglycollate-induced peritonitis, including lymphocytes, granulocytes, and monocytes/macrophages (Figure 1A). Compatible with previous studies,14,22,28 total leukocytes (Live/Single/CD45+ cells) (Figure 1B) and monocytes/macrophages (Live/Single/CD45+/CD11b+/CD11cLow-Med/Ly6G– cells) (Figure 1C) in the lavage fluid of plasminogen-deficient (hereafter Plg−/−) mice were significantly reduced compared with the wild-type littermates. This diminution has previously been shown to be due to impaired recruitment of circulating monocytes to the peritoneal cavity. The monocyte/macrophage population could be divided into 2 groups (labeled large peritoneal macrophages and small peritoneal macrophages [SPMs] in Figure 1D) depending on the differential expression level of Ly6C, CD11b, CD86, and F4/80, as previously described.41 The large peritoneal macrophage population displayed CD11bHi, Ly6CMed, F4/80Hi, and CD86Hi expression, whereas the SPM population was CD11bLow, Ly6CLow, F4/80Low, and CD86Low. Cell numbers in both populations (Figure 1E-F) were reduced upon thioglycollate stimulation in Plg−/− mice compared with Plg+/+ mice, albeit to a lesser degree in the SPM population.

Plasminogen promotes macrophage and dendritic cell accumulation at sites of inflammation. (A) FSC vs SSC plots for all genotypes represent all cell populations. Flow cytometry analysis of peritoneal lavage fluid at 72 hours after intraperitoneal thioglycollate injection shows decreased accumulation of total hematopoietic cells (B) and monocytes/macrophages (C) in the peritoneal cavity of plasminogen-deficient mice. The left panels in B and C are examples of flow cytometry of lavage fluid from individual mice. (D) Macrophages displayed differential expression levels of CD11b, Ly6C, CD86, and F4/80 markers. Thioglycollate stimulation significantly affected the large peritoneal macrophage (LPM) population (E), whereas the SPM population was mildly affected (F). Data are shown as individual values, with means and standard errors indicated.
Plasminogen promotes macrophage and dendritic cell accumulation at sites of inflammation. (A) FSC vs SSC plots for all genotypes represent all cell populations. Flow cytometry analysis of peritoneal lavage fluid at 72 hours after intraperitoneal thioglycollate injection shows decreased accumulation of total hematopoietic cells (B) and monocytes/macrophages (C) in the peritoneal cavity of plasminogen-deficient mice. The left panels in B and C are examples of flow cytometry of lavage fluid from individual mice. (D) Macrophages displayed differential expression levels of CD11b, Ly6C, CD86, and F4/80 markers. Thioglycollate stimulation significantly affected the large peritoneal macrophage (LPM) population (E), whereas the SPM population was mildly affected (F). Data are shown as individual values, with means and standard errors indicated.
Elimination of fibrinogen restores macrophage migration in plasminogen-deficient mice
The impaired recruitment of macrophages to the peritoneum after thioglycollate injection was previously linked to the retention of macrophages within the peritoneal wall.22 Interestingly, histologic analysis of anterior abdominal walls of Plg−/− mice 72 hours after induction of peritonitis revealed a markedly increased fibrin(ogen) accumulation compared with wild-type littermates (Figure 2A-B,D). Staining of anterior abdominal wall from thioglycollate-injected fibrinogen-deficient mice revealed the specificity of the antibody (Figure 2C-D).

Fibrin(ogen) accumulates in the peritoneal wall of plasminogen-deficient mice after peritoneal thioglycollate injection. Immunofluorescence staining of the peritoneal wall of Plg+/+ mice (A), Plg−/− littermates (B), and Fib−/− mice (C) for fibrin(ogen) 72 hours after peritoneal thioglycollate injection (40×). (D) Mean AI555 intensity representing fibrin accumulation was quantified in 5 fields per mouse. Data are shown as individual values with means indicated. DAPI, 4′,6-diamidino-2-phenylindole.
Fibrin(ogen) accumulates in the peritoneal wall of plasminogen-deficient mice after peritoneal thioglycollate injection. Immunofluorescence staining of the peritoneal wall of Plg+/+ mice (A), Plg−/− littermates (B), and Fib−/− mice (C) for fibrin(ogen) 72 hours after peritoneal thioglycollate injection (40×). (D) Mean AI555 intensity representing fibrin accumulation was quantified in 5 fields per mouse. Data are shown as individual values with means indicated. DAPI, 4′,6-diamidino-2-phenylindole.
Based on these findings, we next sought to determine if fibrin accumulation secondary to impaired fibrinolysis contributes to the diminished migration of macrophages in response to thioglycollate. For this purpose, we challenged cohorts of plasminogen and fibrinogen double-deficient (Plg–/Fib–) mice and their plasminogen and fibrinogen-sufficient (Plg+/Fib+), fibrinogen-deficient (Plg+/Fib–), and plasminogen-deficient (Plg–/Fib+) littermates with peritonitis. These experiments were performed independently in 2 laboratories (NIH and CCRF). Figure 3A represents distribution of lymphocytes, granulocytes, and monocytes/macrophages in Plg+/−/Fib+/−, Plg−/−/Fib+/−, and Plg−/−/Fib−/− mice. In both laboratories, Plg–/Fib+ mice showed the expected, significant diminution in total leukocytes and in macrophages in the peritoneal lavage fluid, compared with Plg+/Fib+ littermates (Figure 3B-E). Interestingly, both laboratories independently observed that fibrinogen deficiency (Plg−/−/Fib−/−) rescued the reduced recruitment of monocytes/macrophages imposed by plasminogen deficiency (Plg−/−/Fib+/−). Of note, one laboratory (CCRF) observed a significant increase in leukocyte and macrophage migration in the Plg+/Fib– mice compared with Plg+/Fib+ littermates (Figure 3D-E). At the NIH, there was a trend toward increased migration in the Plg+/Fib– animals that did not reach statistical significance.

Loss of fibrin(ogen) restores macrophage migration in plasminogen-deficient mice. (A) FSC vs SSC plots for all genotypes represent all cell populations. Flow cytometry analysis of peritoneal lavage fluid at 72 hours after thioglycollate stimulation shows normalized recruitment of total hematopoietic cells (B) and macrophages (C) to the peritoneal cavity. The left panels in B and C are examples of flow cytometry of lavage fluid from individual mice. (D) Independent experiment analyzing cell accumulation in peritoneal lavage fluid at 72 hours after thioglycollate stimulation. Plg−/−/Fib+/+ mice exhibited the expected decrease in leukocyte migration. Plg+/+/Fib−/− mice had a significant increase in leukocyte migration after peritonitis induction. Deletion of both plasminogen and fibrinogen returned the phenotype to the wild-type migration pattern. (E) Examination of cytospins from the peritoneal lavage fluid confirmed that the total leukocyte count was driven by the predominant cell type, macrophages. Data are shown as individual values with means indicated. n.s., not significant.
Loss of fibrin(ogen) restores macrophage migration in plasminogen-deficient mice. (A) FSC vs SSC plots for all genotypes represent all cell populations. Flow cytometry analysis of peritoneal lavage fluid at 72 hours after thioglycollate stimulation shows normalized recruitment of total hematopoietic cells (B) and macrophages (C) to the peritoneal cavity. The left panels in B and C are examples of flow cytometry of lavage fluid from individual mice. (D) Independent experiment analyzing cell accumulation in peritoneal lavage fluid at 72 hours after thioglycollate stimulation. Plg−/−/Fib+/+ mice exhibited the expected decrease in leukocyte migration. Plg+/+/Fib−/− mice had a significant increase in leukocyte migration after peritonitis induction. Deletion of both plasminogen and fibrinogen returned the phenotype to the wild-type migration pattern. (E) Examination of cytospins from the peritoneal lavage fluid confirmed that the total leukocyte count was driven by the predominant cell type, macrophages. Data are shown as individual values with means indicated. n.s., not significant.
Because fibrin(ogen) was a significant determinant of macrophage migration in the absence of plasminogen, we examined fibrinogen content of the peritoneal lavage fluid after induction of peritonitis (supplemental Figure 1). Interestingly, there was no significant difference in the fibrinogen content of the peritoneal lavage fluid, as documented by either enzyme-linked immunosorbent assay or western blot analysis. However, western blot analysis did reveal an increase in the higher molecular weight species of fibrin in the wild-type animals compared with the Plg–/Fib+ animals. These data imply that in wild-type animals, plasmin liberates fibrin degradation products that are able to be evaluated on a western blot. However, in the absence of plasmin(ogen), no fibrin degradation products are liberated. This finding further supports our hypothesis that fibrinolysis in the wild-type mice facilitates macrophage migration.
Plasminogen is required for macrophage migration through fibrin
To further test our hypothesis that fibrin(ogen) engagement constitutes the primary impediment to monocyte/macrophage migration in plasminogen-deficient mice, we used an in vitro transwell migration assay. Specifically, we measured the ability of BMDMs or RAW 264.7 cells to cross a fibrin matrix in response to complement factor C5a. In the absence of fibrin, BMDMs and RAW 264.7 cells readily migrated through the membrane with or without plasminogen. Addition of plasminogen to RAW 264.7 cells or BMDMs resulted in a significant 1.7- and 2.3-fold increase in migration, respectively (Figure 4A-B), consistent with a previous in vivo report.18 We next assessed the migration capability of BMDMs or RAW 264.7 cells through a cross-linked fibrin polymer–coated membrane in the transwell chamber. Interestingly, the BMDMs and RAW 264.7 cells essentially did not migrate through a fibrin matrix, with <1% of the transwell surface area covered by macrophages (Figure 4G-H). However, in the presence of plasminogen, both RAW 264.7 cells and BMDMs were able to migrate, with a 212- and 13-fold increase in migration, respectively (Figure 4I-J).
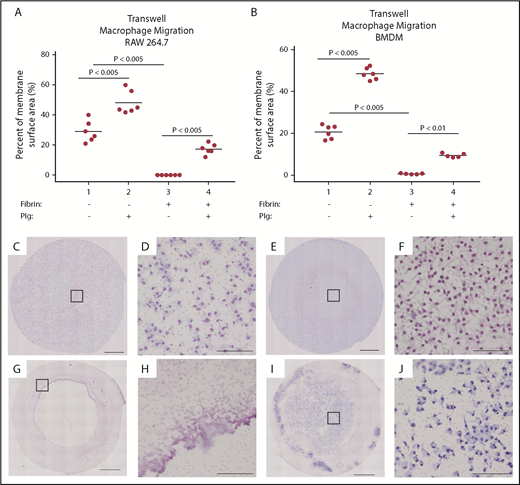
Macrophages require plasminogen to transverse a fibrin matrix in vitro. RAW 264.7 cells (A) and BMDMs (B) were plated into uncoated transwell membranes (1 and 2) or into transwell membranes onto which fibrin had been polymerized (3 and 4) and induced to migrate with C5a in the absence (1 and 3) or presence (2 and 4) of plasminogen. A significant increase in the cellular migration was noted with plasminogen alone (1 vs 2), consistent with previous reports. Cells were essentially unable to migrate in the presence of fibrin without plasminogen (3). However, addition of plasminogen to the cells permitted migration in response to C5a (4). (C-J) Representative images of transwell membranes after overnight incubation with or without fibrin matrix; low power imaging (10×) with higher power magnification (as noted by square outline). Panels D, F, H, and J show the higher power imaging of the transwell membrane. In the absence of fibrin, cells readily migrated without (C-D) or with (E-F) plasminogen. In the presence of fibrin, essentially no cellular migration was noted in the absence of plasminogen (G-H). However, addition of plasminogen restored migration (I-J). Scale bars in panels C, E, G, and I indicate 1 mm. Scale bars in D, F, H, and J indicate 100 µm.
Macrophages require plasminogen to transverse a fibrin matrix in vitro. RAW 264.7 cells (A) and BMDMs (B) were plated into uncoated transwell membranes (1 and 2) or into transwell membranes onto which fibrin had been polymerized (3 and 4) and induced to migrate with C5a in the absence (1 and 3) or presence (2 and 4) of plasminogen. A significant increase in the cellular migration was noted with plasminogen alone (1 vs 2), consistent with previous reports. Cells were essentially unable to migrate in the presence of fibrin without plasminogen (3). However, addition of plasminogen to the cells permitted migration in response to C5a (4). (C-J) Representative images of transwell membranes after overnight incubation with or without fibrin matrix; low power imaging (10×) with higher power magnification (as noted by square outline). Panels D, F, H, and J show the higher power imaging of the transwell membrane. In the absence of fibrin, cells readily migrated without (C-D) or with (E-F) plasminogen. In the presence of fibrin, essentially no cellular migration was noted in the absence of plasminogen (G-H). However, addition of plasminogen restored migration (I-J). Scale bars in panels C, E, G, and I indicate 1 mm. Scale bars in D, F, H, and J indicate 100 µm.
Plasminogen is not required for macrophage migration through a fibrin matrix lacking the integrin αMβ2-binding motif
One mechanism through which fibrin(ogen) directly interacts with leukocytes is through an integrin αMβ2-binding motif, present on the γ chain of fibrinogen. This interaction provides an important local cue for leukocytes in settings of infection and inflammation.23,26,42 We hypothesized that fibrin provided an obstacle to macrophage migration in the absence of plasminogen either by constituting a physically impenetrable barrier or, alternatively, by the “sequestration” of macrophages via the engagement of the myeloid integrin αMβ2-binding motif. To test this theory, RAW 264.7 cells or BMDMs were plated on a fibrin matrix formed with either wild-type mouse fibrinogen or with fibrinogenγ390-396A, which lacks the integrin αMβ2-binding motif (Figure 5A-B).26 As was noted earlier, RAW 264.7 cells and BMDMs were essentially unable to migrate through the wild-type fibrin matrix in the absence of plasminogen. In the absence of plasminogen, again <1% of the membrane surface area was covered by cells (Figure 5C-D). However, in the presence of plasminogen, both cell lines migrated readily (27.5% and 9.9%, respectively) (Figure 5E-F). In sharp contrast, both RAW 264.7 cells and BMDMs readily migrated through the fibrin matrix formed from fibrinogenγ390-396A in the absence of plasminogen (46- and 138-fold increase over wild-type fibrinogen) (Figure 5G-H). The addition of plasminogen to both RAW 264.7 cells and BMDMs also significantly increased migration through fibrin composed of fibrinogenγ390-396A (3.4- and 1.9-fold) (Figure 5I-J).
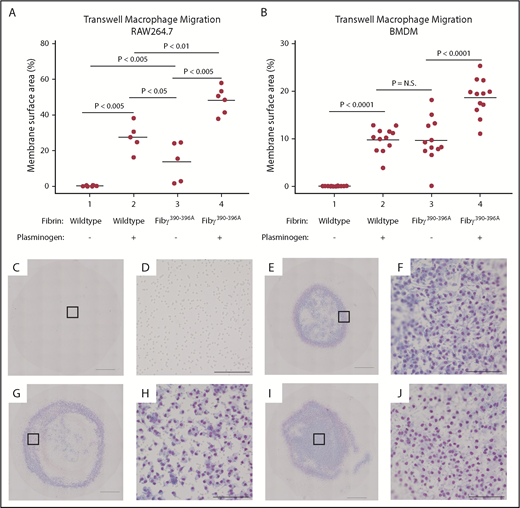
Elimination of the fibrinogen integrin αMβ2-binding motif negates the requirement of plasminogen for macrophage migration through fibrin matrices. Neither RAW 264.7 cells (A) nor BMDMs (B) migrate through wild-type fibrinogen in the absence of plasminogen (Column 1) that is restored in the presence of plasminogen (Column 2). However, neither cell type requires plasminogen to migrate through fibrin matrices if the integrin αMβ2-binding site is abrogated (Column 3). A further increase in migration, even in the absence of the αMβ2-binding motif, is seen with the addition of plasminogen (Column 4). (C-J) Representative transwell membranes from wild-type fibrinogen without (C-D) or with (E-F) plasminogen show migration only in the presence of plasminogen; low power imaging (10×) with higher power magnification (as noted by square outline). Panels D, F, H, and J exhibit higher power imaging of the transwell membrane. Macrophages readily migrated without (G-H) or with (I-J) plasminogen if fibrin was formed from Fibγ390-396A plasma. Scale bars in panels C, E, G, and I indicate 1 mm. Scale bars in panels D, F, H, and J indicate 100 µm. N.S., not significant.
Elimination of the fibrinogen integrin αMβ2-binding motif negates the requirement of plasminogen for macrophage migration through fibrin matrices. Neither RAW 264.7 cells (A) nor BMDMs (B) migrate through wild-type fibrinogen in the absence of plasminogen (Column 1) that is restored in the presence of plasminogen (Column 2). However, neither cell type requires plasminogen to migrate through fibrin matrices if the integrin αMβ2-binding site is abrogated (Column 3). A further increase in migration, even in the absence of the αMβ2-binding motif, is seen with the addition of plasminogen (Column 4). (C-J) Representative transwell membranes from wild-type fibrinogen without (C-D) or with (E-F) plasminogen show migration only in the presence of plasminogen; low power imaging (10×) with higher power magnification (as noted by square outline). Panels D, F, H, and J exhibit higher power imaging of the transwell membrane. Macrophages readily migrated without (G-H) or with (I-J) plasminogen if fibrin was formed from Fibγ390-396A plasma. Scale bars in panels C, E, G, and I indicate 1 mm. Scale bars in panels D, F, H, and J indicate 100 µm. N.S., not significant.
Fibrin(ogen)-mediated impairment of leukocyte migration in plasminogen-deficient mice requires the myeloid integrin αMβ2-binding motif
To test the hypothesis that fibrinolysis is required to liberate macrophages from αMβ2-dependent sequestration to allow effective migration, we used previously described Fibγ390-396A mice, which express a form of fibrinogen lacking the myeloid integrin αMβ2-binding site.26 These mice have normal hemostatic function; however, they express fibrin(ogen) that is unable to interact with αMβ2. We interbred Plg−/− mice with Fibγ390-396A mice, and the generated littermates were challenged with thioglycollate-induced peritonitis. Notably, in this line of experimentation, Plg+/− and Fibγ390-396A/WT (hereafter referred to as FibγHET) were used as controls for the Plg−/− and Fibγ390-396A animals, respectively. At 72 hours after thioglycollate injection, peritoneal lavage fluid was collected and analyzed by using flow cytometry (at NIH)/cytospin (at CCRF). FSC vs SSC plots represent distribution of all cell populations (Figure 6A). As expected, at 72 hours after thioglycollate injection, plasminogen-deficient Plg−/−/FibγHET mice displayed significantly reduced recruitment of total leukocytes (Figure 6B) and monocytes/macrophages (Figure 6C) compared with plasminogen-sufficient Plg+/−/FibγHET control mice. Interestingly, total leukocyte and macrophage recruitment was completely restored in plasminogen-deficient mice by elimination of the αMβ2-binding motif from fibrinogen, as shown by comparing Plg−/−/Fibγ390-396A and Plg+/−/FibγHET control mice. Fibrin(ogen) staining of the anterior abdominal wall of thioglycollate-injected mice showed increased fibrin(ogen) accumulation in Plg−/− and Plg−/−/Fibγ390-396A mice (Figure 6D). Moreover, F4/80 staining of the abdominal wall sections revealed an accumulation of monocytes/macrophages in the areas surrounding blood vessels in Plg−/− mice compared with Plg−/−/Fibγ390-396A and Plg+/+ mice (Figure 6E-F).
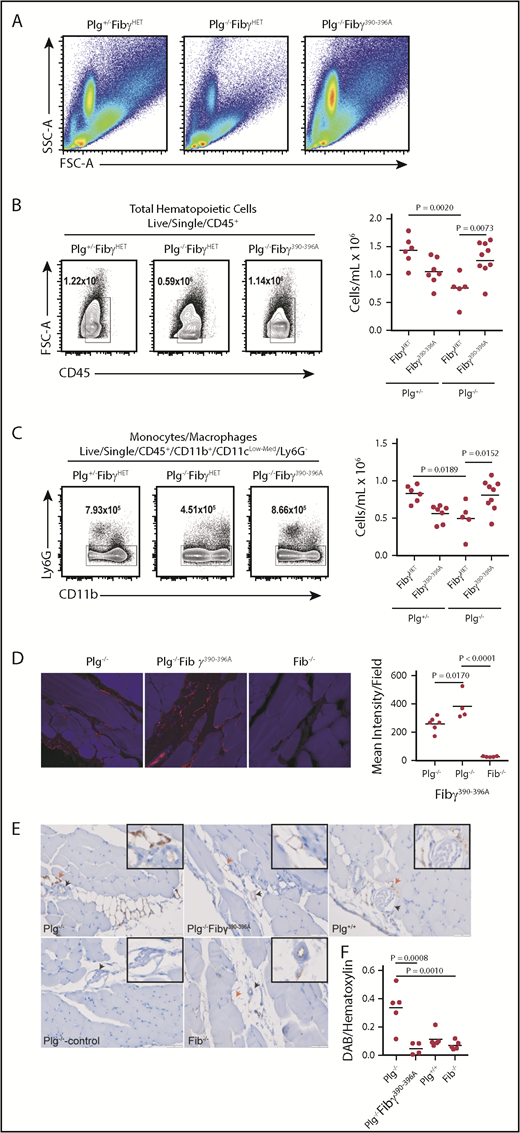
The integrin αMβ2-binding motif on fibrin(ogen) impairs macrophage migration in plasminogen-deficient mice. (A) FSC vs SSC plots for all genotypes represent all cell populations. Flow cytometry analysis of peritoneal lavage fluid at 72 hours after thioglycollate stimulation. Impaired recruitment of total CD45+ hematopoietic cells (B) and monocytes/macrophages (C) in plasminogen-deficient mice was rescued by homozygosity for the Fibγ390-396A allele. The left panels in B and C are examples of flow cytometry of lavage fluid from individual mice. (D) Representative images of immunofluorescence staining of the peritoneal wall of Plg−/− (left), Plg−/−Fibγ390-396A mice (middle), and Fib−/− mice (right) for fibrin(ogen) at 72 hours after thioglycollate injection (40×). Quantitation of fibrin accumulation on the right. (E) Representative images of immunohistochemistry of the peritoneal wall of Plg−/− mice (top left) and Plg−/−Fibγ390-396A mice (top middle) and Plg+/+ mice (top right) of macrophage (F4/80) staining at 72 hours after thioglycollate injection (40×). Secondary antibody only (Plg−/−, bottom left) and Fib−/− (bottom middle) mice were used as controls. Arrowheads indicate blood vessels (black) and macrophage accumulation (red). (F) Quantitation of macrophage accumulation. Quantitation data are shown as individual values with means indicated.
The integrin αMβ2-binding motif on fibrin(ogen) impairs macrophage migration in plasminogen-deficient mice. (A) FSC vs SSC plots for all genotypes represent all cell populations. Flow cytometry analysis of peritoneal lavage fluid at 72 hours after thioglycollate stimulation. Impaired recruitment of total CD45+ hematopoietic cells (B) and monocytes/macrophages (C) in plasminogen-deficient mice was rescued by homozygosity for the Fibγ390-396A allele. The left panels in B and C are examples of flow cytometry of lavage fluid from individual mice. (D) Representative images of immunofluorescence staining of the peritoneal wall of Plg−/− (left), Plg−/−Fibγ390-396A mice (middle), and Fib−/− mice (right) for fibrin(ogen) at 72 hours after thioglycollate injection (40×). Quantitation of fibrin accumulation on the right. (E) Representative images of immunohistochemistry of the peritoneal wall of Plg−/− mice (top left) and Plg−/−Fibγ390-396A mice (top middle) and Plg+/+ mice (top right) of macrophage (F4/80) staining at 72 hours after thioglycollate injection (40×). Secondary antibody only (Plg−/−, bottom left) and Fib−/− (bottom middle) mice were used as controls. Arrowheads indicate blood vessels (black) and macrophage accumulation (red). (F) Quantitation of macrophage accumulation. Quantitation data are shown as individual values with means indicated.
As was observed in one laboratory (CCRF) with the Fib−/− mice (Figure 3D-E), Fibγ390-396A mice exhibited a statistically significant, 1.5-fold increase in leukocyte migration into the peritoneal cavity at 72 hours after induction of peritonitis compared with their wild-type littermates (Figure 7A). Examination of cytospins of the lavage fluid confirmed that this increase in leukocyte migration was driven by a significant, 1.5-fold increase in macrophage migration (Figure 7B). To confirm that this difference between the 2 laboratories was due to the difference in controls used, FibWT, FibγHET, and Fibγ390-396A mice were challenged with thioglycollate-induced peritonitis (supplemental Figure 3). Fibγ390-396A animals again exhibited a significant increase in both total leukocyte and macrophage migration (1.5- and 1.6-fold, respectively) compared with FibWT mice. However, FibγHET animals exhibited an intermediate phenotype between the FibWT and Fibγ390-396A animals. Because factor XIIIa–mediated binding to fibrinogenγ390-396A is modestly diminished,43 we investigated peritoneal macrophage migration in factor XIII–deficient (FXIII−/−) mice. Unlike the Fibγ390-396A mice, total leukocyte and macrophage migration to the peritoneal cavity in FXIII−/− mice in response to thioglycollate injection was indistinguishable from that of FXIII+/+ control mice.

Macrophage migration is dependent on the fibrinogen αMβ2-binding motif but not on factor XIII–mediated fibrin crosslinking. Mice expressing a form of fibrinogen that is unable to interact with the integrin αMβ2 display increased total leukocyte counts (A) and macrophage (B) migration in sterile peritonitis. This increased migration is not seen in mice that are unable to crosslink fibrin (FXIII−/−). (C) In vitro migration of macrophages through fibrin was examined in the presence or absence of factor XIIIa. In the absence of plasminogen, macrophages were unable to migrate through a fibrin matrix regardless of the degree of factor XIIIa-mediated crosslinking (lanes A-D). In contrast, in the presence of plasminogen, macrophages are able to migrate through fibrin regardless of the degree of factor XIIIa–mediated crosslinking (lanes E-H).
Macrophage migration is dependent on the fibrinogen αMβ2-binding motif but not on factor XIII–mediated fibrin crosslinking. Mice expressing a form of fibrinogen that is unable to interact with the integrin αMβ2 display increased total leukocyte counts (A) and macrophage (B) migration in sterile peritonitis. This increased migration is not seen in mice that are unable to crosslink fibrin (FXIII−/−). (C) In vitro migration of macrophages through fibrin was examined in the presence or absence of factor XIIIa. In the absence of plasminogen, macrophages were unable to migrate through a fibrin matrix regardless of the degree of factor XIIIa-mediated crosslinking (lanes A-D). In contrast, in the presence of plasminogen, macrophages are able to migrate through fibrin regardless of the degree of factor XIIIa–mediated crosslinking (lanes E-H).
Macrophage migration in vitro is not dependent on factor XIII
Fibrinγ390-396A does have a modest impairment in factor XIIIa–mediated crosslinking compared with FibrinWT.43 Therefore, to assess if the effect observed in the fibrinogenγ390-396A experiment was related to fibrin crosslinking, macrophage migration was observed with varying concentrations of factor XIIIa added (from 0-5 µg/mL). Even in the complete absence of factor XIIIa–mediated crosslinking, essentially no migration was observed (<1% of the membrane surface area) in the absence of plasminogen (Figure 7C, lanes A-D). However, after plasminogen was added, a significant increase in macrophage migration was observed at all factor XIIIa concentrations used (from 33- to 46-fold increase), compared with identical conditions without plasminogen (Figure 7C, lanes A-D vs E-H). In the presence of plasminogen, factor XIIIa–mediated crosslinking also did not affect macrophage migration, with similar migration with all concentrations of factor XIIIa used (Figure 7C, lanes E-H).
Discussion
A critical role of plasminogen in macrophage migration is now well established.18,28,30-35 Plasmin is a promiscuous enzyme with many potential targets that may affect macrophage migration. Here, we demonstrate for the first time that macrophage migration in inflammation is highly dependent on fibrinolysis. We found that fibrinogen deficiency restores macrophage migration in plasminogen-deficient mice. In addition, the peritoneal lavage fluid from Plg– animals showed a decrease in high-molecular-weight fibrin species compared with that of Plg+ mice after peritonitis. This outcome is expected because in the absence of plasmin, minimal high-molecular-weight fibrin species would be liberated from a fibrin matrix. This further supports the concept that degradation of the fibrin matrix by plasmin is permissive of macrophage migration. Furthermore, macrophages were unable to cross a fibrin matrix in vitro in the absence of plasminogen. This fibrin(ogen)-mediated impairment of macrophage migration in both cases was linked to macrophage engagement of the integrin αMβ2-binding motif on the γ chain of fibrin(ogen). Thus, elimination of the αMβ2-binding motif from fibrin(ogen) rescued macrophage migration in mice lacking plasminogen, and plasminogen was not required for macrophage migration through a fibrin matrix lacking the αMβ2-binding motif. We further confirmed that this mechanism is related to αMβ2 engagement and not loss of factor XIIIa binding to fibrinogenγ390-306A or altered factor XIIIa crosslinking, as FXIII−/− animals had no change in macrophage migration in response to thioglycollate-induced peritonitis. Although there was a significant migration increase in mice lacking the αMβ2-binding motif, this increase was not present in mice unable to crosslink the fibrin polymer.
The key data presented in this article were generated independently in laboratories at 2 different institutions, thus strengthening the rigor of these observations. One difference that was noted between the observations at NIH and CCRF concerned macrophage migration in Fib−/− and Fibγ390-396A mice. No difference in macrophage migration in Plg+/Fib+ mice vs Plg+/Fib– mice was observed at the NIH. An increase in macrophage migration in the Plg+/Fib– mice, compared with Plg+/Fib– mice, was observed at CCRF. However, these data do not necessarily conflict, as there were 2 key differences in the experimental design. At the NIH, Fib−/− mice were compared with Fib+/− littermates, whereas at CCRF, the control mice were Fib+/+. The heterozygous Fib+/− mice (a knockout of the fibrinogen α chain) express 70% of plasma fibrinogen levels.36 This decrease in fibrinogen levels has been noted to be biologically significant in at least one other context.16 Similarly, at CCRF, an increase in migration was noted in the Fibγ390-396A mice compared with FibWT mice. At the NIH, increased migration was not observed. However, in this instance, the control mice used were FibγHET littermates. In FibγHET mice, one would expect ∼75% of the fibrinogen molecules to contain at least one fibrinogenγ390-396A chain and 25% of the fibrinogen molecules to consist entirely of fibrinogenγ390-396A. We have further confirmed that the FibγHET animals have an intermediate macrophage migration phenotype between FibWT and Fibγ390-396A mice. Indeed, there was a statistically significant increase in macrophage migration in the FibγHET animals compared with FibWT animals. In addition, at the NIH, both the Fib+/− and FibγHET control mice were used in conjunction with a Plg+/− genotype. Plg+/− mice also have an intermediate phenotype between Plg+/+ and Plg−/− mice.28
This study is the first to show that fibrinolysis is required for macrophage migration. Fibrin(ogen) deposited at sites of inflammatory disease lesions is a fundamental signal for the innate immune system. The fibrin(ogen) contribution to inflammatory diseases ranges from the immune response in infection to cancer progression and neuroinflammatory disease.23,39,44-47 In many of these settings, loss of fibrin(ogen) is beneficial for disease progression. However, our data suggest that the impact of fibrin(ogen) on physiological or disease processes may be contextual, in terms of timing and location. Indeed, we recently showed that plasminogen deficiency delayed the onset of demyelinating disease in mice, somewhat paradoxically given the known role of fibrin(ogen) in neuroinflammation.17 However, the simultaneous loss of fibrinogen abrogated the delay of demyelinating disease onset. Although there may be fundamental differences in experimental setting, our present findings are consistent with our previous findings in neuroinflammatory disease. Previously, leukocyte migration was examined in Fib−/− and Fibγ390-396 animals in response to thioglycollate.42 No differences were detected compared with wild-type controls in these experiments. However, in the previous findings, leukocyte migration was assessed at 5 hours after thioglycollate injection and not at maximal macrophage accumulation at 72 hours.28,42
Our data implicate the macrophage interaction with the αMβ2-binding motif of fibrin(ogen) as a blockade to further macrophage migration in the absence of plasminogen. Indeed, a fibrin matrix composed entirely of fibrinogenγ390-396A was not a barrier to migration in vitro. This is consistent with previous data that fibrin(ogen) is a crucial local cue for leukocytes. In peritonitis induced by Staphylococcus aureus, fibrin(ogen) is required for clearance of bacteria from the peritoneal cavity and is dependent on the αMβ2-binding motif.26,39 Similarly, in models of neuroinflammation, presence of the αMβ2-binding motif is a key signal that prompts demyelination.23 Therefore, plasmin-mediated macrophage disengagement from fibrin(ogen) seems to be necessary to allow macrophages to continue to migrate to sites of infection or inflammation. It is possible that plasmin-mediated fibrinolysis is also the event that allows macrophages to disengage from inflammatory sites, thus allowing tissue healing to occur. Supporting this hypothesis, absent fibrinolysis significantly impairs wound healing,48 which is then normalized by the absence of fibrin(ogen).37 Our findings do not specifically indicate αMβ2 as the macrophage integrin involved in impeding macrophage migration through fibrin. The αMβ2-binding motif on the fibrinogen γ chain also interacts with αX (CD11c).49,50
Plasmin(ogen) is required to be localized on the macrophage cell surface for normal migration.30,32-35,51 Elimination of plasminogen receptors on macrophages results in a phenotype that is indistinguishable from plasminogen deficiency in terms of macrophage migration. Localization of plasmin activity to the macrophage surface is not only important for migration, however. Elimination of fibrin deposits is also critically dependent on both plasmin and macrophage activity.52 Macrophages are unable to clear fibrin deposits if lacking plasmin activity. Thus, fibrinolysis is critical in both macrophage migration and for macrophages to resolve extracellular fibrin deposits.
Other possibilities for the mechanism of abnormal macrophage migration in the setting of absent fibrinolysis are certainly possible. Formation of an impassable fibrin matrix barrier may also be a mechanism driving the requirement for fibrinolysis. Furthermore, factor XIIIa–mediated crosslinking of fibrin in the Fibγ390-396A mice differs in biologically meaningful ways from fibrin in FibWT mice.43 We therefore assessed macrophage migration in mice unable to crosslink the fibrin polymer. Although we had observed increased macrophage migration in Fibγ390-396A mice compared with FibWT, no similar increase was observed in the FXIII−/− mice, suggesting that a crosslinked polymer was not a requirement for impeding macrophage migration in this experimental setting. We further showed that macrophage migration is not dependent on factor XIIIa–mediated crosslinking in vitro.
The macrophage migration defect caused by plasminogen deficiency has been previously attributed to deficits in MMP9 activation. Other studies have also reported that PAR-1 plays a role in plasminogen-dependent macrophage migration.18,22 Our study does not exclude MMP9 or PAR-1 activation as being a contributor to macrophage migration; however, it does definitively show that plasmin-dependent fibrinolysis is a requirement for effective macrophage migration in the experimental setting of sterile peritonitis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Mary-Jo Danton and Nicholas William Ashton for critically reviewing the manuscript.
This work was funded in part from the intramural program of the National Institutes of Health, National Institute of Dental and Craniofacial Research (NIDCR) (T.H.B. and N.M.M.) and by the CCHMC Gap Funding (E.S.M.) and ASH Bridge Award (E.S.M.). This research was also made possible through use of the NIDCR Combined Technical Core (ZIC DE000729-09) and the NIDCR Veterinary Resource Core (ZIC DE000740-05).
Authorship
Contribution: L.M.S., T.H.B., and E.S.M. designed and planned the experiments with input from N.M.M.; L.M.S., A.G.L., C.T., M.W.S., Z.G., and E.S.M. performed the experiments; L.M.S. and A.G.L. analyzed and discussed the data; M.J.F. and N.M.M. provided strategic guidance throughout the project; L.M.S., M.W.S., T.H.B., and E.S.M. wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: E.S.M. has received honoraria and served on advisory boards for Shire and OctaPharma for topics unrelated to this work. The remaining authors declare no competing financial interests.
Correspondence: Eric S. Mullins, Division of Hematology, Cancer and Blood Diseases Institute, Cincinnati Children’s Research Foundation, MLC 7015, 3333 Burnet Ave, Cincinnati, OH 45229-3039; e-mail: eric.mullins@cchmc.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal