

Key Points
Gene expression identifies DHITsig+ DLBCL tumors with BCL2 and MYC translocations detectable by whole-genome sequencing, but not by breakapart FISH.
Additional genetic mechanisms of MYC dysregulation include focal MYC and MIR17HG copy-number gains and PVT1 promoter deletions.

Abstract
High-grade B-cell lymphomas with MYC and BCL2 and/or BCL6 rearrangements (HGBL-DH/THs) include a group of diffuse large B-cell lymphomas (DLBCLs) with inferior outcomes after standard chemoimmunotherapy. We recently described a gene expression signature that identifies 27% of germinal center B-cell DLBCLs (GCB-DLBCLs) as having a double-hit–like expression pattern (DHITsig) and inferior outcomes; however, only half of these cases have both MYC and BCL2 translocations identifiable using standard breakapart fluorescence in situ hybridization (FISH). Here, 20 DHITsig+ GCB-DLBCLs apparently lacking MYC and/or BCL2 rearrangements underwent whole-genome sequencing. This revealed 6 tumors with MYC or BCL2 rearrangements that were cryptic to breakapart FISH. Copy-number analysis identified 3 tumors with MYC and 6 tumors with MIR17HG gains or amplifications, both of which may contribute to dysregulation of MYC and its downstream pathways. Focal deletions of the PVT1 promoter were observed exclusively among DHITsig+ tumors lacking MYC translocations; this may also contribute to MYC overexpression. These results highlight that FISH fails to identify all HGBL-DH/THs, while revealing a range of other genetic mechanisms potentially underlying MYC dysregulation in DHITsig+ DLBCL, suggesting that gene expression profiling is more sensitive for identifying the biology underlying poor outcomes in GCB-DLBCL.
Introduction
Diffuse large B-cell lymphomas (DLBCLs) are a genetically heterogeneous group of neoplasms with variable outcomes after standard chemoimmunotherapy (eg, R-CHOP [rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone]). There is an ongoing interest in delineating subgroups within DLBCL that share targetable biology, with the potential for precision medicine to overcome biology currently associated with treatment failure. Among the subgroups with the worst outcomes, high-grade B-cell lymphomas with MYC and BCL2 (and sometimes BCL6) rearrangements (HGBL-DH/TH-BCL2s) mostly belong to the germinal center B-cell (GCB) molecular subgroup (the subgroup generally considered to have favorable prognosis).1,2 We recently defined a gene expression signature that identifies nearly one-third of GCB-DLBCL tumors as having a double-hit–like gene expression pattern (DHITsig), with only half harboring rearrangements of both MYC and BCL2 as determined by breakapart fluorescence in situ hybridization (FISH).3 In that cohort, the outcomes for the 20 non–HGBL-DH/TH-BCL2 patients were comparable to those of patients with HGBL-DH/TH-BCL2.
In contrast to Burkitt lymphoma (BL), where the partner of MYC is universally an immunoglobulin gene, in approximately half of HGBL-DH/THs with DLBCL morphology, the partner is nonimmunoglobulin.4 For this reason, a breakapart FISH strategy is favored for detecting MYC rearrangements in DLBCL rather than MYC/IGH dual fusion. Breakapart FISH purportedly fails to detect 2% to 4% of MYC rearrangements when the false-negative cases are identified using an MYC/IGH dual-fusion approach.5-7 Recently, a BL tumor was described where the coding exons of MYC (8.6 kb) were inserted into the IGH locus, an event cryptic to both FISH approaches.8 This raises the possibility that a proportion of DLBCLs harbor MYC and/or BCL2 rearrangements that are cryptic to FISH.
We hypothesized that genetic dysregulations of MYC and BCL2 and their downstream signaling pathways are among the fundamental genetic events underlying the DHITsig in most, if not all, DHITsig+ DLBCL tumors. Using whole-genome sequencing (WGS) of 20 tumors lacking MYC and/or BCL2 rearrangements by FISH, we identified 6 cases with rearrangements cryptic to breakapart FISH, along with other genetic mechanisms of dysregulation of MYC and downstream pathways.
Methods
DNA was extracted from the 20 fresh-frozen GCB DHITsig+ tumors lacking MYC and/or BCL2 translocations (breakapart FISH) identified by Ennishi et al.3 Polymerase chain reaction–free libraries were constructed from sheared DNA that was end repaired and A-tailed using the NEBNext Ultra II DNA library prep kit for Illumina (New England Biolabs) before the ligation of dual-indexed TruSeq adapters and cleanup using 0.8X AMPure XP beads (Beckman-Coulter). Libraries were sequenced to an average depth of ×60 on an Illumina HiSeqX system. Reads were aligned using BWA-MEM (version 0.7.6a).9 Structural variants were identified using Manta.10 Copy-number variants (CNVs) were identified with Control-FREEC.11 Copy-number data were obtained for the discovery cohort using Affymetrix SNP6.0 arrays as previously described.1,12 All statistical tests were performed in R software (version 3.5.1). This study was reviewed and approved by the University of British Columbia–BC Cancer Research Ethics Board in accordance with the Declaration of Helsinki.
Results and discussion
FISH-cryptic rearrangements of BCL2 and MYC
All FISH-detected BCL2 and MYC rearrangements were detected by WGS structural variant calling, confirming the sensitivity of WGS to these events (Figure 1A; supplemental Table 1, available on the Blood Web site). Additional BCL2 and MYC rearrangements were identified in the genomes of 3 cases each. Figure 1B describes 1 of 2 tumors in which the BCL2 gene was inserted into the IGH locus, placing BCL2 in close proximity to the Eµ enhancer. The breakpoints in IGH and near the 3′ untranslated region of BCL2 were consistent with typical BCL2-IGH translocations.13 Importantly, the removal of BCL2 from chromosome 18 left the regions targeted by breakapart FISH probes adjacent (supplemental Figure 1). In another case, 80 kb of the IGK locus, including the Eκ enhancer, was inserted telomeric to BCL2 (Figure 1C). Two tumors harbored cryptic enhancer insertions near MYC (eg, 170 kb of the ZCCHC7 locus including the PAX5 superenhancer inserted telomeric to MYC; Figure 1D). None of these enhancer insertions were large enough to separate the breakapart probes so as to be detectable by FISH (supplemental Figure 1).
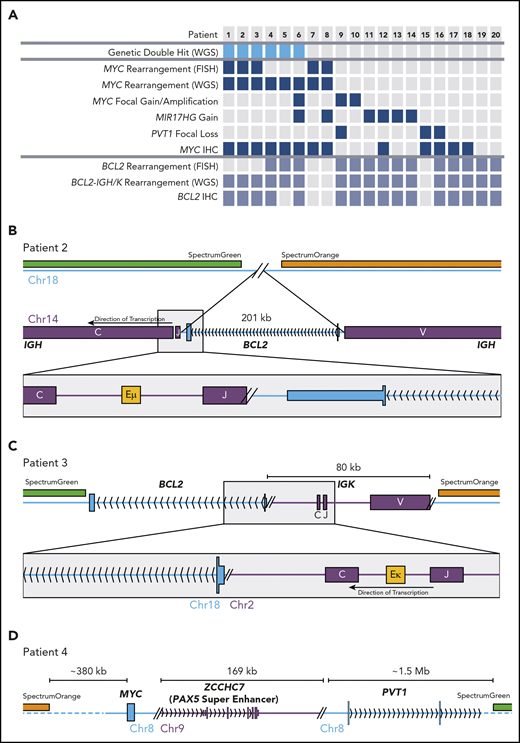
FISH-cryptic MYC and BCL2 rearrangements identified by WGS in DHITsig+ DLBCL tumors. (A) Diagram summarizing the occurrence of each genetic event identified in the 20 non–HGBL-DH/TH-BCL2 DHITsig+ genomes. The thresholds for MYC and BCL2 immunohistochemistry (IHC) positivity were 40% and 50%, respectively. (B-C) Cryptic BCL2 translocations identified in tumors that were negative for BCL2 rearrangement by breakapart FISH. (D) A cryptic MYC rearrangement identified in a tumor that was negative for MYC rearrangement by breakapart FISH.
FISH-cryptic MYC and BCL2 rearrangements identified by WGS in DHITsig+ DLBCL tumors. (A) Diagram summarizing the occurrence of each genetic event identified in the 20 non–HGBL-DH/TH-BCL2 DHITsig+ genomes. The thresholds for MYC and BCL2 immunohistochemistry (IHC) positivity were 40% and 50%, respectively. (B-C) Cryptic BCL2 translocations identified in tumors that were negative for BCL2 rearrangement by breakapart FISH. (D) A cryptic MYC rearrangement identified in a tumor that was negative for MYC rearrangement by breakapart FISH.
Thus, 6 apparently non–HGBL-DH/TH tumors were confirmed to be HGBL-DH/TH through the use of WGS. All of the cases with cryptic BCL2 or MYC rearrangements were positive for BCL2 or MYC protein by immunohistochemistry, respectively (Figure 1A). Cryptic MYC translocations exclusively involved a nonimmunoglobulin partner, whereas BCL2 rearrangements all involved immunoglobulin partner loci. In 2 tumors, the MYC locus was translocated adjacent to the BCL6 superenhancer (supplemental Table 1), an event cryptic to both MYC and BCL6 FISH. No other cryptic BCL6 rearrangements were detected in this patient group. Considering these findings, breakapart FISH failed to detect a clinically significant proportion of HGBL-DH/THs, with cryptic HGBL-DH/TH-BCL2 tumors representing 19% (6 of 31) of all HGBL-DH/TH-BCL2 tumors in the original DHITsig discovery cohort.
CNVs enriched in DHITsig+ tumors
Although focal MYC gains were not exclusive to DHITsig+ tumors, several of the genomes had notable CNVs affecting MYC and/or pathways downstream of MYC, including 2 focal MYC gains and 1 case of double minute (Figure 2A). MIR17HG encodes the miR-17∼92 microRNA cluster, which is a direct target of MYC.14,15 Copy gains affecting MIR17HG were identified in 6 of 20 DHITsig+ cases (Figure 2B) and may represent an avenue for dysregulation of MYC and its downstream targets in these cases.
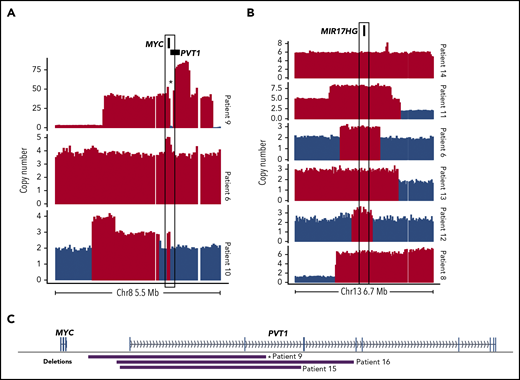
Somatic CNVs affecting MYC, MIR17HG, and PVT1 identified by WGS among DHITsig+ DLBCL tumors. Focal copy-number gains of MYC (A) or MIR17HG (B) identified among DHITsig+ DLBCL tumors. Red bars indicate regions where Control-FREEC identified a significant increase in copy number (P < .05). Compared with 162 GCB tumors without a MIR17HG amplification as determined by single-nucleotide polymorphism (SNP) arrays, expression of MIR17HG was significantly elevated among these 6 cases with MIR17HG copy gains (log2 fold change, 0.85; Wilcoxon P = .032). (C) The boundaries of focal PVT1 transcription start site (TSS) deletions identified in DHITsig+ tumors. *Indicates the PVT1 deletion identified in the double-minute chromosome.
Somatic CNVs affecting MYC, MIR17HG, and PVT1 identified by WGS among DHITsig+ DLBCL tumors. Focal copy-number gains of MYC (A) or MIR17HG (B) identified among DHITsig+ DLBCL tumors. Red bars indicate regions where Control-FREEC identified a significant increase in copy number (P < .05). Compared with 162 GCB tumors without a MIR17HG amplification as determined by single-nucleotide polymorphism (SNP) arrays, expression of MIR17HG was significantly elevated among these 6 cases with MIR17HG copy gains (log2 fold change, 0.85; Wilcoxon P = .032). (C) The boundaries of focal PVT1 transcription start site (TSS) deletions identified in DHITsig+ tumors. *Indicates the PVT1 deletion identified in the double-minute chromosome.
A comparison of SNP array copy-number profiles from 179 GCB-DLBCLs, including the 20 cases described here, revealed that amplifications (copy number >3) of MIR17HG and FCGR2B and deletions of CDKN2A and 22q11.22 (IGL) were significantly enriched among DHITsig+ tumors (supplemental Figure 2). FCGR2B amplification is associated with rituximab resistance and may therefore contribute to treatment resistance among DHITsig+ tumors.12,16 Deletions of CDKN2A lead to decreased TP53 protein17 and represent an additional mechanism of TP53 inactivation in DHITsig+ tumors, along with the frequent TP53 mutations described previously.3 The 22q11.22 deletion breakpoints observed by WGS were consistent with IGL VJ recombination.
A single genome harbored the 11q aberration pattern, which has been described among MYC− BLs.18 The SNP array copy-number data identified 6 cases with the 11q aberration: 1 each in GCB DHITsig+ cases with or without genetic double hit, and 4 in GCB DHITsig− tumors lacking MYC or BCL2 rearrangements. Four 11q aberrations were confirmed by FISH (supplemental Figure 3). These results suggest that the 11q aberration is not associated with the DHITsig in DLBCL.
Among the genomes, there were 3 instances of focal promoter deletions of PVT1, including 1 in the double-minute chromosome (Figure 2C). The SNP array copy-number data did not identify any additional cases with this deletion. The PVT1 long noncoding RNA gene contains intragenic enhancers that drive expression of PVT1, but in the absence of an active PVT1 promoter they instead drive expression of MYC.19 Although the number of occurrences limits our power to determine whether PVT1 deletions are associated with DHITsig positivity, these results suggest that this is a rare, albeit recurrent, event that could promote MYC expression in DHITsig+ tumors.
In summary, genetic events affecting both MYC and BCL2 were identified in 13 of 20 DHITsig+ tumors that were deemed non–HGBL-DH/THs by breakapart FISH. The identification of FISH-cryptic MYC and BCL2 rearrangements in 6 cases highlights the limitation of FISH in identifying HGBL-DH/TH. The DHITsig phenotype can thus be attributed to a range of genetic events affecting MYC, BCL2, and downstream pathways, some of which are not revealed even by WGS, demonstrating that gene expression profiling is a more appropriate method to identify this biological group that constitutes high-risk GCB-DLBCL.
Data will be deposited in EGA with accession number EGAS00001004285.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This research was supported by grants from the Terry Fox Research Institute (1061) and Canadian Institute for Health Research (R.D.M.) and MITACS Accelerate (R.D.M. and L.K.H.), a Scholar Award from the ASH Foundation (R.D.M.), and the BC Cancer Foundation. R.D.M. is a Michael Smith Foundation for Health Research Scholar.
Authorship
Contribution: L.K.H., B.M.G., J.T., M.A., C.K.R., A.J., and R.D.M. analyzed and interpreted the data; L.K.H. generated all figures; M.B., B.M., and S.B.-N. processed the tissues and performed FISH assays; and L.K.H., D.W.S., and R.D.M. conceived the study and wrote the manuscript.
Conflict-of-interest disclosure: A.J., D.W.S. and R.D.M. are named inventors on a patent application pertaining to the DHITsig. The remaining authors declare no competing financial interests.
Correspondence: Ryan D. Morin, Department of Molecular Biology and Biochemistry, Simon Fraser University, 8888 University Dr, Burnaby, BC, Canada; e-mail: rdmorin@sfu.ca; and David W. Scott, Department of Lymphoid Cancer Research, BC Cancer, 675 West 10th Ave, Vancouver, BC, Canada; e-mail: dscott8@bccancer.bc.ca.
