

Key Points
MF (cutaneous T-cell lymphoma) exhibits substantial clonotypic heterogeneity of malignant T cells.
Cutaneous lesions of MF are formed by seeding of clonotypically heterogeneous neoplastic T-cell clones from the blood to the skin.

Abstract
Mycosis fungoides (MF) is a mature T-cell lymphoma currently thought to develop primarily in the skin by a clonal expansion of a transformed, resident memory T cell. However, this concept does not explain the key characteristics of MF, such as the debut with multiple, widespread skin lesions or inability of skin-directed therapies to provide cure. The testable inference of the mature T-cell theory is the clonality of MF with respect to all rearranged T-cell receptor (TCR) genes. Here, we used a whole-exome sequencing approach to detect and quantify TCR-α, β, and γ clonotypes in tumor cell clusters microdissected from MF lesions. This method allowed us to calculate the tumor cell fraction of the sample and therefore an unequivocal identification of the TCR clonotypes as neoplastic. Analysis of TCR sequences from 29 patients with MF stage I to IV proved the existence of multiple T-cell clones within the tumor cell fraction, with a considerable variation between patients and between lesions from the same patient (median, 11 clones; range, 2-80 clones/sample). We have also detected multiple neoplastic clones in the peripheral blood in all examined patients. Based on these findings, we propose that circulating neoplastic T-cell clones continuously replenish the lesions of MF, thus increasing their heterogeneity by a mechanism analogous to the consecutive tumor seeding. We hypothesize that circulating neoplastic clones might be a promising target for therapy and could be exploited as a potential biomarker in MF.
Introduction
Cutaneous T-cell lymphomas (CTCLs) are mature T-cell neoplasms, among which mycosis fungoides (MF) is the most common disease entity.1 MF presents initially as scaly, erythematous patches and plaques on skin, which may progress to tumors and disseminate to lymph nodes and other organs, such as the central nervous system.2-4 The pathogenesis of MF has been studied for decades as a model disease reflecting key characteristics of low-grade lymphomas such as progressive course and lack of curative treatments.
MF is believed to originate from the mature, memory, tissue-resident T cells expressing skin homing markers cutaneous leucocyte-associated antigen and CCR4.5,6 This straightforward hypothesis explains the affinity of MF to the skin and its low capacity to disseminate to extracutaneous sites. However, some clinical and molecular features of MF are incompatible with the model of the skin-resident memory T cell as the origin of MF. It is unexplainable why the disease usually starts multifocally in different areas of the skin rather than at a single site representing the location of the founding, transformed T cell. Second, even profound depletion of lymphocytes in the skin (eg, by electron beam radiation therapy or psoralen UV A therapy) almost never results in a cure and only provides short-term responses.7-10 Third, cells sharing molecular characteristics of malignant T cells in MF have been found in the bone marrow of the patients years before the emergence of skin lesions of the disease,11 and CTCL can be transmitted via bone marrow transplant from asymptomatic donors.12,13 Fourth, MF may share the common precursor with other lymphomas (eg, Hodgkin lymphoma) that do not originate in the skin but primarily occupy extracutaneous sites, such as lymph nodes.14
These observations are more compatible with a scenario in which CTCL originates by the hematogenous spread of precursor neoplastic cells to the skin niche.15 However, this concept was met with skepticism and resistance, because analysis of the clonality of the T-cell receptor (TCR) seemed to strongly indicate that this disease is monoclonal and originates in the skin.6,16
TCR clonality assays have been the most powerful technique to dissect the pathogenesis of CTCL and other T-cell lymphomas.17,18 During T-cell development, the V, (D), and J gene segments of TCRG, TCRB, and TCRA undergo sequential rearrangements producing unique CDR3 sequences that are retained in mature T cells. The diversity at CDR3 is further increased by insertions or deletions at V(D)J junctions, and therefore, those sequences constitute a unique signature of a given T-cell clone.19 Most research focused on TCRG because of its relatively small size and limited diversity and only recently on TCRB, which is more diverse and has a unique property of allelic exclusion, which simplifies data analysis.
We have recently shown that MF cells sampled from a plaque or a tumor may share the same TCR-γ clonotype but exhibit different TCR-β and TCR-α clonotypes.20,21 Since TCR-γ loci (TCRG) rearrange before TCR-β (TCRB) and TCR-α (TCRA) and the unique TCRG CDR3 sequences are inherited by the T cells derived from those early clones, these findings are incompatible with the current model of the mature T cell as the precursor of MF, where all malignant cells would share an identical clonotype for all rearranged TCR genes (TCRG, TCRB, and TCRA).
Skin-resident T cells do not recirculate but remain in the tissue, where they can survive and proliferate without migration to the lymph nodes.5,22,23 Although atypical malignant T cells are conspicuously absent in the blood in MF patients in the early stages and the results of clonality assays are usually negative, it may be argued that standard TCR-γ detection methods are rather insensitive for detecting rare clones in the background of highly diverse normal T cells.24 Indeed, using more careful experimental approaches such as tumor fraction enrichment by laser capture microdissection and analysis of purified lymphocytes or mononuclear cells from the blood, some authors were able to find clonal, circulating cells even in the early stages of disease development.20,25-28 Unfortunately, due to the difficulties in polymerase chain reaction (PCR) amplification of TCRB and TCRA from genomic DNA, these findings rely heavily on the analyses of TCR-γ, which may give false-positive results because of the low diversity of TCRG. Likewise, relying on reverse transcription PCR–based methods for TCR-β may miss malignant clones with unproductive TCR rearrangements.
Here, we have applied the technique of TCR detection by whole-exome sequencing (WES)21 to revisit the hypothesis of circulating neoplastic cells in MF. By comparing TCR clonotypes in the skin and blood in patients with MF, we reveal a complex pattern of recirculation of tumor subclones. We propose that the substantial clonotypic heterogeneity of skin lesions in MF is caused by the mechanism of consecutive seeding of the skin niche by multiple subclones of neoplastic cells.
Materials and methods
Patients, sample collection, and storage
We included 29 patients with diagnosis of MF established by a certified dermatopathologist or a hematopathologist. Patient characteristics are summarized in supplemental Table S1 (available on the Blood Web site). None of the patients received systemic therapy, radiotherapy, or phototherapy at the time of tissue sampling. Most patients used medium- and high-potency topical steroids, but the biopsy specimens were obtained from the lesions that had not been treated with steroids for at least 3 days. Ethical approval was obtained from the Health Research Ethics Board of Alberta, Cancer Committee HREBA.CC-16-0820-REN1. After informed consent, 4-mm punch skin biopsy specimens were collected from patients and embedded in optimal cutting temperature medium stored at −80°C. Then, 10 mL blood was collected into an EDTA tube, and peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll centrifugation and resuspended in 50% Dulbecco’s modified Eagle medium (catalog number 11965-084; Thermo Fisher Scientific), 40% fetal bovine serum (catalog number 16000044; Thermo Fisher Scientific), and 10% dimethyl sulfoxide (catalog number 20688; Thermo Fisher Scientific) and frozen in liquid nitrogen until further use. Before DNA isolation, the PBMCs were thawed and resuspended in RPMI 1640 medium with 10% fetal bovine serum. DNA and RNA was isolated with Trizol reagent (catalog number 15596026; Invitrogen, Carlsbad, CA).
Cryosectioning, laser capture microdissection, and sample preparation for WES
Skin biopsy specimens were cryosectioned and prepared for WES according to the previously reported protocol (supplemental Figure 7).21 The NEBNext Ultra II DNA library prep kit for Illumina (catalog number E7645S; New England Biolabs) was used to prepare the samples for sequencing, and SSELXT Human All exon V6 +UTR probes (Agilent Technologies) were used for exome capture. The DNA libraries were sequenced on an Illumina HiSeq 1500 sequencer using a paired-end 150 kit (catalog number PE-402-4002, HiSeq paired-end rapid cluster kit V2) or NovaSeq 6000 S4 reagent kit 300 cycles (catalog number 20012866).
Data analysis
The fastq files were analyzed using MiXCR29 (version 2.10.0) to identify the TCR clonotypes. For WES data, partial reads were filtered out, as these might be the captures of only V or J sequences. The reads were processed using the GATK430 (version 4.0.10) generic data-preprocessing workflow and analyzed with Titan31 (version 1.20.1) to determine copy-number aberration and tumor cell fraction using the hg38 human reference genome. The tcR32 package for R was used to calculate the overlapping clones.
Results
TCRB sequencing of MF shows lesional and topological heterogeneity of malignant clonotypes
We have previously provided evidence that WES can be used for identifying neoplastic TCR clonotypes (ie, the TCR CDR3 sequences specific to malignant T cells in MF).21 Our method relies on the sequencing of CDR3 regions and quantification of the fraction of TCR-α, β, and γ clonotypes corresponding to the tumor cell percentage, thus filtering out the TCR sequences from the reactive, tumor-infiltrating T cells. Using the same approach here, we have identified neoplastic clonotypes in 29 patients with MF using microdissected samples of 46 biopsy specimens from cutaneous lesions (Figure 1; supplemental Table 1). To quantify clonogenic heterogeneity, we focused on TCRB, which is sufficiently diverse to avoid the risk identical rearrangements of unrelated T-cell clones and which is rearranged on a single chromosome in >98% of all T cells (allelic exclusion), thus unequivocally defining a T-cell clone.33-36 In a purely monoclonal disease, one can expect that the frequency of the single, dominant TCR-β clonotype matches the tumor cell fraction of the sample. However, in our 46 skin biopsy specimens, the tumor cell fraction significantly exceeded the frequency of the most abundant TCR-β clonotype, which unequivocally proved clonotypic heterogeneity of MF. We identified a range of 2 to 80 TCR-β clonotypes per sample, which corresponded to the tumor cell fraction of the sample (ie, neoplastic clonotypes) (Figure 2A; supplemental Figure 1A). On average, the most frequent (dominant) TCR-β clonotype comprised only 19.32% of the tumor fraction, which was similar to the values obtained in other studies using the PCR-based approach and next-generation sequencing.37,38 The number of neoplastic TCR-β clonotypes correlated with the tumor cell fraction, but not with the stage of the lesion (T1 plaque vs T2 tumor), which further supported the notion that those clonotypes represented true tumor clones and were not derived from infiltrating, reactive T cells (Figure 2B).
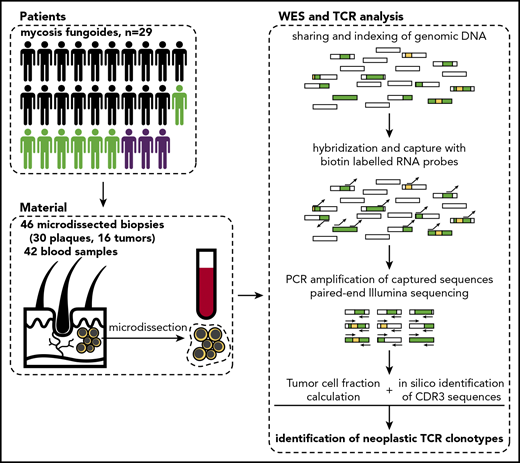
Schematic representation of sample collection and processing. Single or multiple punch biopsy specimens (4 mm) and 10 mL total blood were collected from 29 patients. In 7 patients, we collected >1 biopsy specimen (green silhouettes), and 3 patients were followed longitudinally with several biopsies and/or blood samples. The skin biopsy specimens were cryosectioned and used for laser microdissection of clusters of tumor cells, which along with blood PBMCs were processed for WES. Sequenced data were analyzed to identify rearranged CDR3 sequences of TCRA, TCRB, and TCRG and determine tumor cell fraction. Rectangles represent DNA fragments, green areas represent exons, and yellow areas represent rearranged TCR genes.
Schematic representation of sample collection and processing. Single or multiple punch biopsy specimens (4 mm) and 10 mL total blood were collected from 29 patients. In 7 patients, we collected >1 biopsy specimen (green silhouettes), and 3 patients were followed longitudinally with several biopsies and/or blood samples. The skin biopsy specimens were cryosectioned and used for laser microdissection of clusters of tumor cells, which along with blood PBMCs were processed for WES. Sequenced data were analyzed to identify rearranged CDR3 sequences of TCRA, TCRB, and TCRG and determine tumor cell fraction. Rectangles represent DNA fragments, green areas represent exons, and yellow areas represent rearranged TCR genes.
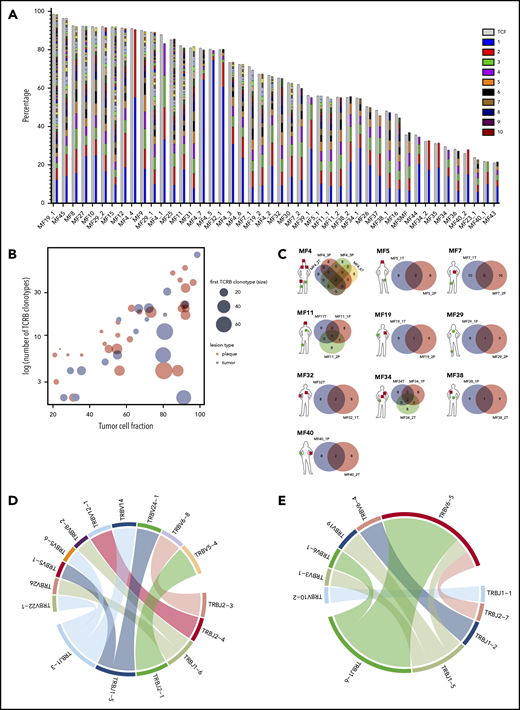
Clonotypic heterogeneity of skin lesions in MF. (A) The TCF for the skin samples was estimated by the copy-number aberration (CNA) data from the WES. The tumor cell fraction (gray bars) is plotted vs the cumulative frequency of the most abundant TCR-β clonotypes. The frequencies of the clonotypes are represented, with stacked bars representing clonotypes from the most abundant (rank 1) to the least frequent. The ranks of clonotypes are color-coded as in the legend. (B) Correlation between tumor cell fraction and the number of neoplastic clonotypes. Note that the clonotypic heterogeneity is not dependent on the stage of the lesion (tumor vs plaque). The size of the circle is proportional to the percentage of the most dominant (rank 1) TCR-β clonotype. (C) Topological heterogeneity in MF. Venn diagrams illustrating the number of overlapping TCR-β clonotypes across different skin lesions. The location and type of the lesion is plotted for each patient (green circle, plaque; red square, tumor). (D-E) VJ combination diversity of TCR-β clonotypes in MF. The combinations of VJ genes of the neoplastic clonotypes are presented in panels D (MF32) and E (MF16).
Clonotypic heterogeneity of skin lesions in MF. (A) The TCF for the skin samples was estimated by the copy-number aberration (CNA) data from the WES. The tumor cell fraction (gray bars) is plotted vs the cumulative frequency of the most abundant TCR-β clonotypes. The frequencies of the clonotypes are represented, with stacked bars representing clonotypes from the most abundant (rank 1) to the least frequent. The ranks of clonotypes are color-coded as in the legend. (B) Correlation between tumor cell fraction and the number of neoplastic clonotypes. Note that the clonotypic heterogeneity is not dependent on the stage of the lesion (tumor vs plaque). The size of the circle is proportional to the percentage of the most dominant (rank 1) TCR-β clonotype. (C) Topological heterogeneity in MF. Venn diagrams illustrating the number of overlapping TCR-β clonotypes across different skin lesions. The location and type of the lesion is plotted for each patient (green circle, plaque; red square, tumor). (D-E) VJ combination diversity of TCR-β clonotypes in MF. The combinations of VJ genes of the neoplastic clonotypes are presented in panels D (MF32) and E (MF16).
In 10 patients, we obtained biopsy specimens from multiple skin lesions, which enabled us to study clonotypic heterogeneity between different areas of the skin (Figure 2C; supplemental Figure 5B-C). In 8 patients, we compared the biopsy specimens from the plaque and the tumor (late lesion) (patients MF4, MF5, MF7, MF11, MF19, MF34, MF38, and MF40), and in 3 patients, we compared lesions in the same stage (2 plaques from MF29, stage T2, and 2 tumors from MF4 and MF32, stage T3). The heterogeneity of any single lesion was lower than the combined heterogeneity of all biopsy specimens from the same patient combined, measured by the clonotype richness (number of different neoplastic TCR-β clonotypes) and Simpson index (the probability that 2 clonotypes, randomly drawn from the sample are different) (supplemental Figure 1). Surprisingly, the degree of overlap clonotypes between different lesions was very low (1-2 clonotypes), and in 1 case (MF7 tumor and plaque), we did not detect any shared clonotypes. Importantly, the shared clonotypes were not always the most frequent ones. Thus, extensive clonotypic heterogeneity was detected not only on the level of a single lesion but also between different lesions (topological heterogeneity).
Neoplastic clonotypes are frequently detected in the peripheral blood in MF
Having established that MF shows high clonotypic heterogeneity, both with regard to the composition of a single lesion and between different lesions, we realized that this heterogeneity could not be generated in the skin in situ, because cells in the MF infiltrate do not express RAG1/2 and terminal deoxynucleotidyltransferase, the key enzymes needed for TCR gene recombination (see Döbbeling et al39 and A.I. and R.G., unpublished observations of RNA sequencing of MF). We considered the possibility that there is a pool of clonotypically heterogeneous neoplastic cells in the circulation that are able to seed the skin and undergo clonal expansion in the skin niche. Therefore, we investigated whether malignant T-cell clones could be found in the peripheral blood. For consistency across samples, we assumed conservatively that the top 10 frequent TCR clonotypes from skin represent the true, tumor-related clonotypes. This assumption is based on the observation that the 10 most abundant TCR-β clonotypes contributed to ≤85% (95% upper confidence interval [CI] value) of the tumor-related clonotypes, whereas the remaining clonotypes (ranked >10) had a low abundance (95% CI, 1.3% to 1.5%) and only contributed to 6% to 15% (95% CI) of the total number of clonotypes. To provide a second layer of validation, we have also compared the TCRA and TCRG CDR3 sequences in the blood and the skin.
In 79% (15/19) of patients, we have detected at least 1 shared TCR-β clonotype between the skin and blood. The same number of patients had ≥1 common TCR-γ shared clonotype in the skin and blood, whereas 16 patients had circulating neoplastic TCR-α clonotypes (Figure 3A; supplemental Figures 2A and 3A). Thus, all patients had ≥1 shared clonotype TCR-α, β, or γ between the skin and blood at the time of sampling. The number of identical clonotypes in the skin and the blood was highest for TCR-α (1-7 clonotypes; supplemental Figure 2A), probably because a single clone of T cells defined by a common TCR-β clonotype may comprise 1 to 3 different TCR-α clonotypes (supplemental Figure 4). Importantly, the neoplastic clonotypes were composed of multiple V-J gene combinations that indicated they originated at the stage of T-cell development (Figure 2D-E).
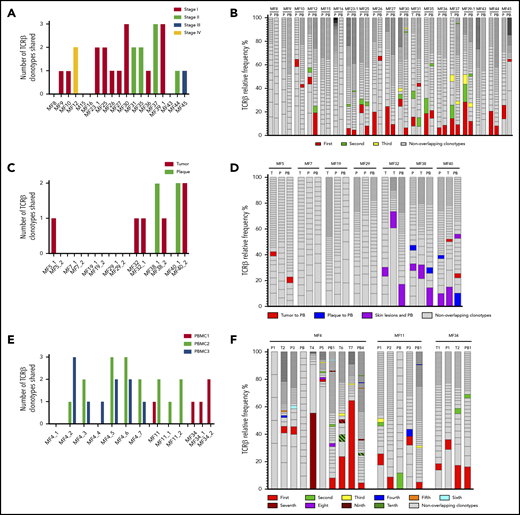
Detection of the neoplastic TCR-β clonotypes in the peripheral blood. The sequences of the TCR-β clonotypes in the blood that matched any of the top 10 neoplastic TCR-β clonotypes identified in the corresponding skin sample were identified to detect the neoplastic clonotypes in the circulation. The number and frequency of those shared neoplastic clonotypes are shown separately for the 3 groups of patients as defined in Figure 1. Nineteen patients with a single biopsy specimen (A-B); patients who had multiple skin biopsies (C-F), of whom in 7 patients, the biopsy specimens were obtained at a single time point (C-D); and 3 patients who were sampled longitudinally (E-F). In panels B, D, and F, the first-ranking shared clonotype in the skin is indicated in red, and the subsequent shared clonotypes are color-coded as indicated in the legend. The nonoverlapping clonotypes are indicated in gray. P, plaque; PB, peripheral blood; T, tumor.
Detection of the neoplastic TCR-β clonotypes in the peripheral blood. The sequences of the TCR-β clonotypes in the blood that matched any of the top 10 neoplastic TCR-β clonotypes identified in the corresponding skin sample were identified to detect the neoplastic clonotypes in the circulation. The number and frequency of those shared neoplastic clonotypes are shown separately for the 3 groups of patients as defined in Figure 1. Nineteen patients with a single biopsy specimen (A-B); patients who had multiple skin biopsies (C-F), of whom in 7 patients, the biopsy specimens were obtained at a single time point (C-D); and 3 patients who were sampled longitudinally (E-F). In panels B, D, and F, the first-ranking shared clonotype in the skin is indicated in red, and the subsequent shared clonotypes are color-coded as indicated in the legend. The nonoverlapping clonotypes are indicated in gray. P, plaque; PB, peripheral blood; T, tumor.
We subsequently analyzed whether the frequency of the clonotypes in the skin correlated with the frequency of those in the blood. The dominant (most abundant) TCR-β clonotype from the skin was identified in the blood in 8 patients, and 6 of those clonotypes were also dominant in the blood (Figure 3B). The pattern was more complicated for TCR-γ and TCR-α, where dominant clonotypes could be detected in the blood and the skin in only 2 patients (MF36 and MF37) (supplemental Figures 2B and 3B).
Similar occurrence of tumor-derived clonotypes was detected in 10 patients in whom we analyzed multiple skin biopsy specimens; paired biopsy specimens (tumor and plaque) were analyzed for 7 patients, and multiple biopsy specimens (between 3 and 7) were analyzed for 3 patients. All patients shared ≥1 clonotype (TCR-β, α, or γ) between the blood and 1 or both skin biopsy specimens (Figure 3C; supplemental Figures 2C and 3C), and the degree of clonotypic sharing was higher between the blood and the skin than between 2 skin biopsy specimens (Figure 3D). No correlation was found between the number of shared clonotypes and the stage of disease and progression-free survival (supplemental Table 1).
It has not escaped our attention that some clonotypes were shared between different samples (both skin and blood) from different patients, such as the CDR3 sequence CDNNNDMRF (TRAV16/TRAJ43), which was found among malignant clonotypes in 8 of 29 patients, and CAASRGC_AKNIQYF (TRBV18/TRBJ2-4), which was found in 20 of 29 patients (supplemental Table 2). We have previously noticed frequent clonotypic sharing between patients with MF and excluded laboratory error as a possible cause.21 We have also excluded the possibility that those sequences represented sequences parts of unrelated captured exomes, with the secondary verification using blastn and blastp that indicated the sequences to be TCR (data not shown).
Temporal dynamics of malignant TCR clonotypes in the skin and blood
Clonotypic heterogeneity could be achieved by seeding the skin with malignant clones, a mechanism that is responsible for the formation of metastases in solid tumors.40-43 Metastatic seeding may occur with single cancer cells (in which case, the metastasis represents a single subclone) or via continuous seeding, when clusters of cancer cells transfer the entire heterogeneity of the primary tumor to the metastases.41-43 The third seeding mechanism, referred to as consecutive seeding, relies on sequential recruitment of neoplastic cells to the metastases and results in metastatic lesions that only represent a fraction of the heterogeneity of the primary tumor (supplemental Figure 6).43 Clonotypic heterogeneity of skin lesions excluded the single-cell seeding events, whereas the fact that we detected single neoplastic clonotypes in the blood rather than combinations of different clonotypes argued against continuous seeding. To further elucidate the mechanism of tumor seeding, we followed the malignant clonotypes in the skin and the blood in 3 patients (MF4, MF11, and MF34) over a period of 9 to 22 months (Figures 3E-F and 4). In each case, we found neoplastic T-cell clonotypes in the blood, defined as those TCRB CDR3 sequences that were found in ≥1 skin biopsy. The number of circulating neoplastic clonotypes varied from 2 clonotypes in MF34 to 10 clonotypes in MF4. Circulating neoplastic clonotypes were not detected constantly in all blood samples, and certain CDR3 sequences could be detected in the blood before occurrence in skin biopsy specimens, such as GPGTRLLVLGERGLLGRGRGR_ WVWFLRGVPGLCSGANVLTF or CASCPH_VSCRRP, which were found in the blood of patient MF4 months before their detection in skin biopsy specimens (Figure 4A). Together with the finding that a single skin lesion contained only a fraction of all possible neoplastic clonotypes, our data strongly supported the model of the development of the skin lesions in MF by consecutive seeding (Figure 5; supplemental Figure 6).
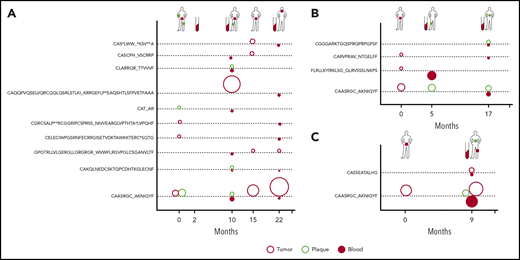
Dynamics of neoplastic clonotypes in the skin and blood. Three patients were followed longitudinally with multiple skin biopsies and/or blood sampling. All shared neoplastic clonotypes are plotted on the time axis for individual patients: MF4 (A), MF11 (B), and MF34 (C). The location and type of lesion are indicated for each patient on a silhouette (green circle, plaque; red square, tumor). Each dotted line corresponds to a single, shared clonotype of the indicated amino acid sequence of CDR3β. Circles above the line are skin clonotypes (open red, tumor; open green, plaque), whereas the solid red circles below the line are the clonotypes detected in the blood. The size of the dot is proportional to the frequency of the shared clonotype in the sample.
Dynamics of neoplastic clonotypes in the skin and blood. Three patients were followed longitudinally with multiple skin biopsies and/or blood sampling. All shared neoplastic clonotypes are plotted on the time axis for individual patients: MF4 (A), MF11 (B), and MF34 (C). The location and type of lesion are indicated for each patient on a silhouette (green circle, plaque; red square, tumor). Each dotted line corresponds to a single, shared clonotype of the indicated amino acid sequence of CDR3β. Circles above the line are skin clonotypes (open red, tumor; open green, plaque), whereas the solid red circles below the line are the clonotypes detected in the blood. The size of the dot is proportional to the frequency of the shared clonotype in the sample.

The hypothesis of tumor seeding in the pathogenesis of MF. Even in the early stages of the disease, patients have circulating neoplastic T-cell clones in peripheral blood. Early lesions are initiated by the pioneer clones and create a niche (blue-shaded area) that facilitates seeding of this area of the skin (lesion 1) with subsequent clones (lesion 2) (consecutive seeding model43 and supplemental Figure 6). The clonal composition of different lesions may differ (lesion 3) due to the stochastic nature of cancer seeding. Some clones may have a higher proliferation capacity in the skin and may overgrow other clones (green clone in lesion 3), further mutate, reenter the circulation (orange arrow), and reseed another area of the skin (lesion 4). The figures represent symbolically the structure of the skin with the epidermis (Epi), dermis (Der), and a pink-shaded blood vessel (BV). Different clones of neoplastic T cells are marked with different colors.
The hypothesis of tumor seeding in the pathogenesis of MF. Even in the early stages of the disease, patients have circulating neoplastic T-cell clones in peripheral blood. Early lesions are initiated by the pioneer clones and create a niche (blue-shaded area) that facilitates seeding of this area of the skin (lesion 1) with subsequent clones (lesion 2) (consecutive seeding model43 and supplemental Figure 6). The clonal composition of different lesions may differ (lesion 3) due to the stochastic nature of cancer seeding. Some clones may have a higher proliferation capacity in the skin and may overgrow other clones (green clone in lesion 3), further mutate, reenter the circulation (orange arrow), and reseed another area of the skin (lesion 4). The figures represent symbolically the structure of the skin with the epidermis (Epi), dermis (Der), and a pink-shaded blood vessel (BV). Different clones of neoplastic T cells are marked with different colors.
Discussion
The major finding of this study is that lesions of MF comprise a highly diverse collection of malignant T-cell clones that percolate between the skin and blood. We propose that consecutive seeding of circulating neoplastic T-cell subclones in the skin is a likely mechanism of growth and evolution of the lesions in this cutaneous lymphoma.
Difficulties in distinguishing between the CDR3 sequences in malignant T cells and those of normal, reactive T cells has been the major limitation of TCR analyses. Several indirect approaches have been used to mitigate this problem, such as setting arbitrary thresholds for clonotypic frequencies or surrogate measures of tumor cell fraction as a ratio of the frequency of the most abundant TCR-β clonotype to the sum of all TCR-β clonotypes.37 Obviously, both approaches assume that all tumor cells are clonal (ie, share a single TCR-β clonotype). This assumption is a cornerstone of the theory of CTCL as a neoplasm of the mature T cell but has not rigorously been tested experimentally. Experimental findings suggesting clonotypic heterogeneity in CTCL44 have either been ignored or attributed to contamination by clonal inflammatory T cells,45 activation of skin resident T cells by superantigens,44 or age-dependent clonal expansion.46-49 We were able to solve this methodological problem by calculating the tumor cell fraction in the sample by WES and thus directly enumerate the clonotypes derived from neoplastic T cells.21 Our method relies on DNA sequencing, which eliminates a potential error due to aberrant TCR expression in cancer cells. With this approach, we confirmed our previous findings20 demonstrating clonal heterogeneity in MF. Our findings contribute to the increasing body of evidence showing that CTCL, as well as other T-cell leukemias and lymphomas, exhibits genomic and transcriptomic heterogeneity, similar to what is seen in solid tumors.50-53
We have found a large variation in the clonotypic richness (number of different TCR-β clonotypes) in MF lesions, ranging from 2 up to 80 distinct clonotypes. In most samples, the frequency of the most abundant TCR-β clonotype is relatively high (19.4%), which is well above the usual 15% threshold accepted by many authors as a hallmark of monoclonality. This is reflected by a relatively low Simpson index (the probability that a random draw from the pool of clonotypes yields 2 different clonotypes; median, 7%; 1st and 3rd quartile, 3.2% to 10.7%) (supplemental Figure 1). Thus, even consecutive draws from the pool of clonotypes of a given lesion are most likely to yield identical clonotypes, which can be misinterpreted as monoclonality (eg, 10 consecutive draws yield identical results in ∼50% of cases). In 33% of biopsy specimens, the frequency of the first TCR-β clone was <10%, which corresponds well to the observed proportion of cases of MF in which monoclonality cannot be detected by standard assays.54
There is compelling evidence that staphylococcal enterotoxins facilitate progression of MF.55 Eradication of Staphylococcus aureus with antibiotics leads to spectacular responses in some cases of MF.56 It is controversial whether superantigens stimulate the neoplastic cells directly or rather indirectly via activation of interleukin-2 by bystander T cells.55,57 Since binding of the superantigen to TCR is efficient for only a small subset of TCR Vβ (TRVB),58 we asked whether those TRVB segments are found in neoplastic cells. In contrast to previous reports,59-61 we did not see any obvious evidence of overrepresentation of superantigen-reactive TRVB segments, although the presence of TRVB5.3, TRVB5.1, TRVB19 was noted in 5 out of 29 patients (supplemental Table 2). Larger studies are needed to determine whether patients expressing those TCR-β variants are more responsive to antibiotic therapy and S aureus elimination.
We have considered potential mechanisms that could explain the finding of clonotypic heterogeneity in absence of a true clonal diversity. We have rejected the idea that clonotypic heterogeneity is attributed to secondary somatic mutations within the already assembled CDR3 region, because we detected that the rearranged TCRB were composed by numerous combinations of various VJ segments. Moreover, extensive secondary mutations within TCRB comes from the quantification of the ratio between TCR-α and TCR-β clonotypes. In a population of normal memory T cells, TCRA/TCRB is between 2 and 3, because TCRA rearrangements are usually Biallelic, and there are occasional secondary rearrangements of this locus.62 Mutations in TCRB would result in an increase in the number of unique TCR-β sequences and thus a decrease in the TCRA/TCRB ratio, which was not the case in our material (the TCRA/TCRB ratio was 2.76). Third, we have detected a higher than expected overlap between clonotypes between patients (supplemental Table 2), which argues against the influence of random mutations. Clonotypic richness of TCR-β would also be increased if malignant T cells were not subject to allelic exclusion of TCRB locus. However, this scenario is unlikely, because allelic exclusion is a stochastic process, which is very resistant to perturbations63,64 and is not violated in other T-cell malignancies investigated to date.52,65 Lack of allelic exclusion would also decrease the TCRA/TCRB ratio in a similar way as described above for secondary mutations. Finally, recombinations of TCRB in postthymic lymphocytes is impossible, because neither normal nor neoplastic T cells in MF express the essential RAG1/2 recombinases.39
Conceptually, the clonotypic heterogeneity described here is different from the mutational subclonal heterogeneity, because it cannot be generated continuously in the tumor and can only be generated continuously in the time span when RAG1/2 recombinases are active (ie, at the level of immature T-cell precursor). It has been suggested though, that multiple malignant clones can be generated from the pool of normal, reactive lymphocytes in the skin undergoing malignant transformation.44 However, such a mechanism would have to operate at an unprecedented efficiency to generate hundreds of cancer clones simultaneously in different areas of the skin and is therefore unlikely. The most likely mechanism is by accrual of malignant T-cell clones from the circulation to the skin. Several lines of indirect evidence support such a hypothesis. First, we were able to detect malignant TCR clonotypes in the blood. It has long been known that TCR-γ clonotypes identical to those in lesional skin could be detected in the blood in MF in 5% to 10% of patients in early-stage disease without any prognostic impact.49,66-68 We have found that the presence of circulating malignant clones is a rule rather than an exception; ≥1 malignant TCR-β or TCR-α clonotype was present in the peripheral blood in all examined patients. Although only a small fraction of the entire pool of neoplastic clonotypes could be detected in the blood, the chances of detecting TCR-β clonotypes increased by repeated sampling, probably because of their very low frequency compared with the background of normal T cells. Second, the neoplastic clonotypes were found in the blood even in the very early stages of the disease, they were not correlated with the stage of the disease, and they did not always represent the dominant clonotype in the skin. Therefore, those circulating clones could not solely represent subclinical leukemization due to disease progression. Third, clonotypic overlap (the number of shared TCR-α, β, or γ clonotypes) was higher between the skin and peripheral blood than between discrete skin lesions.
Taken together, these observations are compatible with the presence of a pool of very diverse neoplastic T-cell clones in the peripheral blood that may seed at a different frequency to the skin, where they develop further into the lesions of MF (Figure 5; supplemental Figure 6). Our findings are compatible with the model of consecutive seeding in which only a fraction of all neoplastic clones transfers the diversity to the developing lesions of lymphoma. Previous modeling of consecutive metastatic seeding events revealed that each lesion is likely to be funded by 10 to 150 cancer clones,43 a number that is in the same range as the 6 to 20 clonotypes (1st-3rd quartiles) detected by us in MF. Although not investigated here directly, we hypothesize that neoplastic clones do not migrate unidirectionally from the blood to the skin. Recent findings that downregulation of CD69 enables skin resident memory cells to exit the tissue and recirculate69 indicate that neoplastic cells in MF should also be able to reenter the circulation and contribute to the pool of circulating neoplastic clones. This mechanism is reminiscent of the phenomenon of tumor self-seeding40,70 where circulating metastatic cells colonize the primary tumor. Tumor self-seeding is enhanced by changes in the tissue niche occupied by cancer, which makes the niche more receptive for the subsequent entry of the waves of circulating tumor cells.70 In MF, the source of those dermotropic neoplastic T-cell clones remains unknown, although there is some evidence that bone marrow may play such a role.11-13 Research showed that the bone marrow niche provides shelter for different types of malignant cells (eg, breast or prostate cancers) and that the bone marrow pool of cancer might be responsible for relapses after therapy.71,72
We wish to highlight several limitations of this study. Sensitivity of WES for TCR analyses is limited, and our method does not detect rare clonotypes. It is therefore possible that we have not captured the entire repertoire of malignant clonotypes. It is also evident that microdissected cells are not representative of the heterogeneity of the entire lesion. It is likely that different tumor clones are not distributed homogeneously throughout the MF lesion and that our sampling method underestimates the true clonal diversity. Finally, our evidence of clonal heterogeneity relies on the statistical analyses of the frequency of different clonotypes in relation to the tumor cell fraction rather than analyses of individual cells. With the emerging technologies allowing for mutation calling and TCR sequencing in single cells,52,73 it should be possible to confirm our findings directly and by independent methods.
The proposed mechanism of tumor self-seeding in the pathogenesis of MF may have several practical implications. Circulating neoplastic T-cell clones may constitute an interesting target for therapy. It is tempting to speculate that mogamulizumab,74 which blocks CCR4, an essential cutaneous homing receptor,75 exerts it therapeutic efficacy via inhibition of skin seeding by circulating cancer clones. Identification of TCR-β clonotypes could also be used diagnostically in CTCL. Interestingly, we have found significant interindividual overlap in TCR-β clonotypic sequences, dramatically exceeding the frequency of shared, public clonotypes in healthy individual. Although the mechanism of clonotypic sharing remains elusive, the common TCRB CDR3 sequences could be easily, quantitatively, and cost-effectively detected with PCR-based techniques and used for the diagnosis and monitoring of therapy in CTCL.
Data have been submitted to the Database of Genotypes and Phenotypes (dbGAP) with accession number phs001877.v1.p1.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge Thomas Salopek, Rachel Doucet, and the nursing staff of Edmonton Kaye Clinic for their help in sample collection. Hanne Fogh provided excellent care to the patients from the Copenhagen Center and helped to collect clinical data. Vibeke Pless and Pia Eriksen helped with the collection and shipment of samples.
This study was supported by grants from the Canadian Dermatology Foundation (CDF RES0035718), the University of Alberta, University Hospital Foundation (University of Alberta), a Bispebjerg Hospital (Copenhagen, Denmark) unrestricted research grant (R.G.), and the Danish Cancer Society (Kræftens Bekæmpelse R124-A7592 Rp12350).
Authorship
Contribution: A.I. designed the experiments, analyzed the data, wrote the manuscript, and submitted data to the Database of Genotypes and Phenotypes; A.I. and S.O. performed the experiments; J.P. and D.H. performed copy-number aberration analysis and tumor cell fraction calculations; W.W. and G.K.-S.W. provided input regarding the technical aspects of the experiments and bioinformatic pipelines and edited the manuscript; R.G. supervised the experiments and data analysis, prepared the visual abstract, and edited the manuscript; and all authors approved the final version of this paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Aishwarya Iyer, Department of Medicine, University of Alberta, 260 HMRC, 114 St and 85th Ave, Edmonton, AB T6G 2R3, Canada; e-mail: aiyer2@ualberta.ca.
