


Abstract
Tumor cells rewire metabolic pathways to adapt to their increased nutritional demands for energy, reducing equivalents, and cellular biosynthesis. Alternations in amino acid metabolism are 1 modality for satisfying those demands. Amino acids are not only components of proteins but also intermediate metabolites fueling multiple biosynthetic pathways. Amino acid–depletion therapies target amino acid uptake and catabolism using heterologous enzymes or recombinant or engineered human enzymes. Notably, such therapies have minimal effect on normal cells due to their lower demand for amino acids compared with tumor cells and their ability to synthesize the targeted amino acids under conditions of nutrient stress. Here, we review novel aspects of amino acid metabolism in hematologic malignancies and deprivation strategies, focusing on 4 key amino acids: arginine, asparagine, glutamine, and cysteine. We also present the roles of amino acid metabolism in the immunosuppressive tumor microenvironment and in drug resistance. This summary also offers an argument for the reclassification of amino acid–depleting enzymes as targeted therapeutic agents.
Introduction
Amino acids perform critical metabolic functions. They are used for the synthesis of proteins, the formation of other low-molecular-weight compounds, and energy production by intermediate metabolites fueling other biosynthetic pathways. They have traditionally been classified as nutritionally essential (indispensable) or nonessential (dispensable) for animals and humans.1 Within the nutritionally essential amino acids, “essential” refers to those whose carbon skeletons cannot be synthesized de novo; “semiessential” refers to those that can be synthesized de novo but not in amounts necessary to maintain human health, necessitating dietary supplementation. Among 20 proteogenic amino acids, 9 are “essential” in humans by that definition: phenylalanine, valine (Val), threonine, tryptophan, methionine, leucine, isoleucine, lysine, and histidine (His). Six are “semiessential:” arginine (Arg), cysteine (Cys), glycine, glutamine (Gln), proline (Pro), and tyrosine. Nonessential amino acids are those that can be synthesized de novo in adequate amounts: aspartate (aspartic acid [Asp]), asparagine (Asn), glutamate (glutamic acid; Glu), alanine, and serine (Ser).2
For the maintenance of hematopoietic stem cells (HSCs), the essential amino acid Val is indispensable, and depletion of Val decreases the number of native HSCs.3 On the other hand, the altered metabolic wiring of leukemic cells enables survival in the metabolically stressful leukemic microenvironment.4 That reprogrammed leukemic cell metabolome plays an important role in tumor growth and chemoresistance. Targeting such metabolic vulnerability, specifically the differences in amino acid dependency between HSCs and leukemia stem cells (LSCs), may unveil novel, targeted leukemia therapies with acceptable therapeutic windows.
The stress responses triggered by amino acid loss are observed in both LSCs and HSCs. Even normal homeostatic processes generate low-level stress and activation of the integrated stress response (ISR) pathway. Activation of the ISR results in eIF2α phosphorylation-dependent upregulation of activating transcription factor 4 (ATF4) that facilitates survival of both HSCs and LSCs.5 However, leukemia cells tend to require a greater supply of nutrients for growth and proliferation than normal cells,6,7 and the leukemia environment is characterized by lower levels of glucose, Gln, and oxygen compared with the normal hematopoietic microenvironment.8
The process of rewiring metabolism to meet increased demands for energy, reducing equivalents, and cellular building blocks often results in cancer cells developing auxotrophy: the inability to synthesize sufficient quantities of amino acids due to dependence on the extracellular pool of amino acids. In contrast, normal cells exhibit lower demand for amino acids.9 That difference establishes a metabolic vulnerability in malignant cells, providing a rationale for amino acid–depletion therapy. Given that leukemia is a heterogeneous clonal disorder originating from LSCs,10 it is important to note that genomically heterogeneous LSC subpopulations propagate in the bone marrow (BM) microenvironment and likely exhibit different metabolic patterns and responses to amino acid–targeting therapies.11
Fueled in part by advances in protein-engineering technology and improved understanding of the metabolic differences between cancer and normal cells, the full potential of amino acid–depletion therapy is starting to be realized9 ; for instance, l-asparaginase has produced excellent results in the treatment of acute lymphoblastic leukemia (ALL) and is a component of the standard of care.12
This review describes novel aspects of amino acid metabolism in leukemia cells and deprivation strategies using heterologous or engineered enzymes that have been successfully used in the treatment of hematopoietic malignancies. We focus on 4 key amino acids, Arg, Asn, Gln, and Cys (Figure 1), along with the roles of amino acid metabolism in the immunosuppressive tumor microenvironment and in drug resistance. We also present the case for reclassifying amino acid–targeting therapies as targeted therapies against LSCs, which require specific amino acid–metabolic pathways for survival.
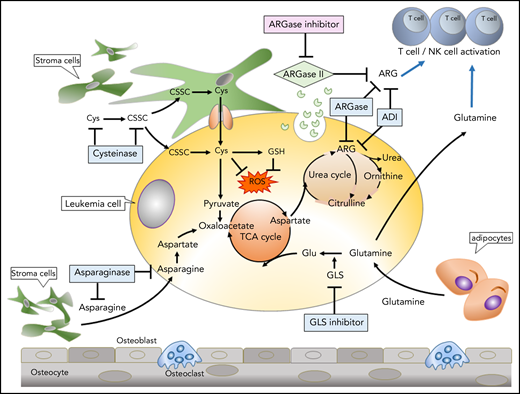
Therapeutic targeting of amino acids in tumors and the tumor microenvironment. The most common amino acid–depletion strategies include depletion of Asn with asparaginase (ASNase), Arg with arginase (ARGase), or Arg deiminase (ADI), Glu with glutaminase (GLS) inhibitors, and Cys/cysteine (CSSC) with cysteinase. Stromal cells or adipocytes produce Asn and Glu, supporting tumor metabolism. ARGase secretion by tumor cells (which depletes extracellular Arg) and/or tumor consumption of Gln may deprive immune cells of these nutrients, and metabolic inhibitors of ARGase or GLNase reverse immune suppression. GSH, glutathione; NK, natural killer; ROS, reactive oxygen species; TCA, the citric acid.
Therapeutic targeting of amino acids in tumors and the tumor microenvironment. The most common amino acid–depletion strategies include depletion of Asn with asparaginase (ASNase), Arg with arginase (ARGase), or Arg deiminase (ADI), Glu with glutaminase (GLS) inhibitors, and Cys/cysteine (CSSC) with cysteinase. Stromal cells or adipocytes produce Asn and Glu, supporting tumor metabolism. ARGase secretion by tumor cells (which depletes extracellular Arg) and/or tumor consumption of Gln may deprive immune cells of these nutrients, and metabolic inhibitors of ARGase or GLNase reverse immune suppression. GSH, glutathione; NK, natural killer; ROS, reactive oxygen species; TCA, the citric acid.
Arginine
Arg has been classified as a semiessential amino acid because healthy individuals can synthesize Arg from other amino acids, such as Gln, Glu, and Pro.13 In pathologic conditions, however, the human body becomes dependent on Arg intake.14 In addition, Arg serves as an intermediate or a substrate in many biological pathways, such as the urea cycle, TCA cycle, and thymotropic activities.15,16 Arg is also the major precursor for synthesis of cancer-associated compounds such as polyamines, nitric oxide (NO), and NO synthetase itself.16,,-19 Previous studies have shown that low NO concentrations promote cancer cell proliferation and angiogenesis, whereas high NO concentrations lead to DNA damage and cell death.20,,,-24
As cancer cells require nutrients to sustain growth, exhausting endogenous sources of Arg is a potential therapeutic intervention. That strategy was successfully demonstrated to be effective in previous studies using polyethylene glycol–conjugated (PEGylated) forms of ADI (ADI-PEG20) or ARGase (PEG-ARGase), the 2 critical enzymes of the Arg metabolism/urea cycle.23,,-26 Figure 2 illustrates the Arg-metabolism cycle. Several studies have shown the beneficial effects of Arg degradation to citrulline by ADI in preventing proliferation of tumor cells such as melanoma, lung cancer, renal cell carcinoma, and hepatocellular carcinoma.27,28 In those experiments, researchers noted similarities among the malignancies, including deficiencies in OTC or arginosuccinate synthetase-1 (ASS1) (http://www.proteinatlas.org). Treatment with ADI depleted the endogenous Arg reservoirs of tumor cells because the cells failed to replace Arg by hydrolyzing citrulline.29
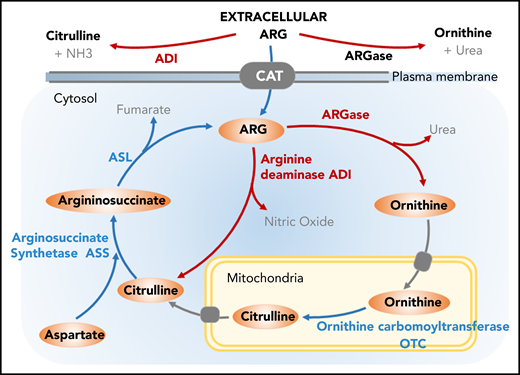
Targeting Arg metabolism. Arg can be synthesized from other amino acids via urea cycle; however, tumor cells lacking the urea cycle enzyme arginosuccinate synthetase (ASS) rely on exogenous supply of Arg. The endogenous sources of Arg can be used as a potential therapeutic intervention in treating cancer. ADI catalyzes Arg degradation to citrulline and ammonium. ARGase generates ornithine as an intermediate, and ornithine carbomoyltransferase (OTC) catalyzes the conversion of ornithine to citrulline. Upon condensation of Asp and citrulline by the enzymatic activity of ASS, arginosuccinate intermediate is generated followed by the reformation of Arg.
Targeting Arg metabolism. Arg can be synthesized from other amino acids via urea cycle; however, tumor cells lacking the urea cycle enzyme arginosuccinate synthetase (ASS) rely on exogenous supply of Arg. The endogenous sources of Arg can be used as a potential therapeutic intervention in treating cancer. ADI catalyzes Arg degradation to citrulline and ammonium. ARGase generates ornithine as an intermediate, and ornithine carbomoyltransferase (OTC) catalyzes the conversion of ornithine to citrulline. Upon condensation of Asp and citrulline by the enzymatic activity of ASS, arginosuccinate intermediate is generated followed by the reformation of Arg.
One limitation in using ADI for cancer therapy is that ADI provokes an immune response in human cells because the enzyme is originally isolated from prokaryotes, specifically Mycoplasma arginini. Furthermore, the short half-life of the native drug (4 hours) places a restraining factor on its efficacy in cancer treatment.24,25 ADI-PEG20 was developed to diminish the drug immunogenicity and to extend the half-life of the enzyme to 6 days.25
Several studies have tested the effect of ADI-PEG20 monotherapy in melanoma and advanced hepatocellular carcinoma; despite the transient drop of Arg levels observed, tumor progression was sustained and a neutralizing antibody was detected in the patients’ plasma.30,,-33 Additional resistance mechanisms that resulted from ADI-PEG20 treatment were autophagy and overexpression of the ASS1 protein. Notably, blocking autophagy with chloroquine in combination with ADI increased the rate of apoptosis in sarcoma cells in vitro, and treating sarcoma samples expressing low levels of ASS1 with ADI-PEG20 reduced cancer proliferation.34,35
Many attempts have been made to combat the resistance that develops to ADI-PEG20 monotherapy. For instance, Beddowes et al proposed that ADI-PEG20 would act synergistically in combination with cisplatin and pemetrexed in thoracic cancer. Their results showed a sustainable drop in Arg level and a significant decrease in the production of neutralizing antibodies.36
A recent study of the efficacy of ADI-PEG20 in combination with cytarabine in treating acute myeloid leukemia (AML) in mouse models and in vitro demonstrated that the combination significantly reduced AML cell percentage and plasma Arg level.37 ADI-PEG20–resistant AMLs showed higher expression of ASS1 and argininosuccinate lyase than ADI-PEG20–sensitive AMLs, whereas ADI-PEG20–sensitive AMLs showed greater Arg uptake from the extracellular milieu via high-affinity cationic amino acid transporter-1 (CAT1), consistent with the Arg-auxotrophic nature of ADI-PEG20–sensitive AMLs. These findings lend support to the suggestion that ADI-PEG20–resistant AMLs enhance the endogenous production of Arg through the ASS1 pathway.37
The other Arg-depleting strategy is ARGase, a catabolic enzyme that converts Arg to ornithine in the urea cycle.38 Mammals express 2 isoforms of this enzyme, ARGase I and ARGase II, which have 60% similarity in their amino acid sequence. ARGase I is a cytosolic enzyme in the human liver and ARGase II is a mitochondrial enzyme in other tissues that is thought to play no role in the urea cycle.39,40 Like ADI, recombinant ARGase has been pegylated to extend its half-life from a few hours to 3 to 4 days. PEG-ARGase treatment diminished the proliferation of OTC- and ASS1-deficient cancer cells, unlike ADI-PEG20, which inhibited only tumors lacking ASS1 expression.38 Mussai et al41 demonstrated the antiproliferative effect of a pegylated human recombinant ARGase (BCT-100) on AML blasts. The mechanism involved the ARG-auxotrophic nature of AML blasts (which constitutively express the Arg transporters, CAT1, and CAT2B) that mediate extracellular Arg uptake. BCT-100 treatment led to rapid depletion of extracellular and intracellular Arg reservoirs, resulting in decreased proliferation of AML blasts.
Depleting Arg in tumor cells by using PEG-ARGase in combination with other drugs has been a focus of various studies; as 1 example, a recent study indicated the antitumor potential of PEG–ARGase I and 5-fluorouracil in hepatocellular carcinoma in vivo.38 Additionally, it has been reported that PEG–ARGase I in combination with cytarabine is an effective approach in treating AML and ALL both in vivo and in vitro.41,42 Despite the great efficacy of that combination, both studies failed to achieve durable responses and resulted in the development of resistance.41,42 PEG–ARGase I treatment decreased the expression of cyclin D3, a fundamental protein in T-cell ALL tumor proliferation, and consequently induced apoptosis in T-cell ALL cell lines isolated from mouse models.42 Furthermore, pegylated human recombinant ARGase impeded T-cell proliferation in ALL in vivo.42
Asparagine and glutamine
Asn is classified as a nonessential amino acid, meaning that it can be synthesized by human cells.43 In glycoprotein synthesis, the amide nitrogen of Asn residues in proteins provides a site of glycosylation to the carbohydrate chain to form the N-linkage.44 Additionally, Asn shows exchange-factor capacity to coordinate the uptake of extracellular Arg, His, and Ser, and it can modulate ammonia metabolism.44,45 The capacity of Asn to suppress apoptosis is evident by its ability to negatively modulate endoplasmic reticulum stress and translation-dependent apoptosis.46
Asn starvation is a potential strategy to induce apoptosis in cancer cells that express a low level or are deficient in Asn synthetase (ASNS).47 Examples of the tumors lacking ASNS are leukemia and lymphoma, which depend on external sources of Asn.48,49 Expression of ASNS has been found to be positively correlated with poor prognosis in selected human tumors.46 Evidence of Asn auxotrophy was first discovered through the antitumor effect of asparaginase (ASNase) in guinea pig serum on lymphoma cells.50,-52 Later, ASNase was isolated from Escherichia coli to treat lymphoblastic leukemia.53
ASNase monotherapy has been an effective approach to treat ALL and some cases of lymphomas, and the combination of vincristine and prednisone with ASNase has resulted in a remission rate of >90% in children with ALL.54,55 In breast cancer, a synergistic interaction between ASNase and doxorubicin in induction of apoptosis has been reported.56 ASNase and temozolomide triggered a synergistic antitumor effect in the growth of subcutaneous gliomas in vivo.57
Although the majority of pediatric ALL patients achieve plasma levels of ASNase that deplete Asn effectively,58 some patients exhibit apparent pharmacokinetic/pharmacodynamic inadequacies.59,-61 Resistance to ASNase is currently thought to follow 1 of 4 patterns, as follows. First, normal and leukemic lymphoblasts may eliminate the ASNase enzyme through immune responses triggered by recognition of ASNase as a foreign protein.61 This results in the secretion of a neutralizing anti-ASNase antibody and blocks the drug activity.62,63 Second, leukemia blasts that rapidly upregulate ASNS protein expression may develop resistance to ASNase.64,65 Third, enhanced production of Gln synthetase and increased activity of Gln transporters (SLC1A5, SLC3A2, SLC38A2, SLC38A3, SLC38A4, SLC38A5) in cancer cells is believed to contribute to ASNase resistance, which can be resolved by inhibiting Gln synthetase.66 Fourth, BM stromal cells, an important element in the cancer microenvironment, release Asn when leukemia blasts are treated with ASNase.67 Previous studies showed that BM mesenchymal stem cells (MSCs) mediate resistance to most chemotherapy reagents. Because MSCs show sensitivity to cytoskeleton-targeting drugs,68 administration of vincristine prior to ASNase treatment has been proposed to impede the MSC-mediated resistance of ALL blasts.69
It has been further reported that ASNase treatment depletes Gln, which can induce apoptosis because many cancer cells require large quantities of Gln to maintain TCA cycle anaplerosis and support cell survival.70 Interestingly, Asn supplementation was reported to be necessary and sufficient to suppress Gln withdrawal–induced apoptosis without restoring the levels of other nonessential amino acids or TCA-cycle intermediates.
Gln depletion by the glutaminase (GLNase) activity of ASNase has been associated clinically with toxicities such as acute pancreatitis, thrombotic disorders, and immunosuppressive effects71,,-74 (Figures 3 and 4). One approach that has been suggested to solve this issue is to isolate or engineer Glu with GLS-free ASNase.75,76 Nevertheless, ASNase purified from bacteria triggers hypersensitivity or anaphylaxis.76,77 E coli expresses 2 isoforms of ASNase: ASNase I and ASNase II. The former is a cytosolic protein that has a low affinity to Asn, and the latter is a periplasmic enzyme that is commonly used in cancer therapy because of its high affinity to Asn.78 In a comparison of PEG–E coli ASNase and Erwinia chrysanthemi ([Erw] now Dickeya dadantii) ASNase (ERWase), E coli ASNase exhibited lower immunogenicity than Erw ASNase, whereas Erw ASNase resulted in less toxicity and is a preferred treatment of patients who are hypersensitive to E coli ASNase.79,-81
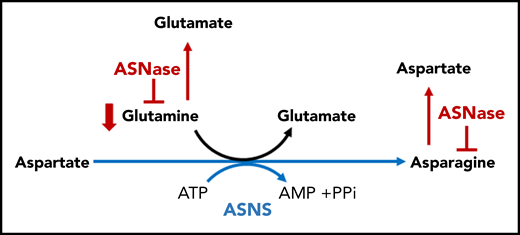
Therapeutic targeting of Asn and Gln. Asn is catalyzed by ASNS, the primary mediator of resistance to ASNase. ASNase hydrolyzes ASN into Asp and ammonia. Asn starvation by ASNase is a potential strategy of apoptosis induction in leukemia and lymphoma cells lacking ASNS and depend on the exogenous supply of Asn. The secondary activity present in ASNase is an GLS activity, which drives hydrolysis of Gln to Glu and ammonia. AMP, adenosine monophosphate; ATP, adenosine triphosphate; PPi, proton pump inhibitor.
Therapeutic targeting of Asn and Gln. Asn is catalyzed by ASNS, the primary mediator of resistance to ASNase. ASNase hydrolyzes ASN into Asp and ammonia. Asn starvation by ASNase is a potential strategy of apoptosis induction in leukemia and lymphoma cells lacking ASNS and depend on the exogenous supply of Asn. The secondary activity present in ASNase is an GLS activity, which drives hydrolysis of Gln to Glu and ammonia. AMP, adenosine monophosphate; ATP, adenosine triphosphate; PPi, proton pump inhibitor.
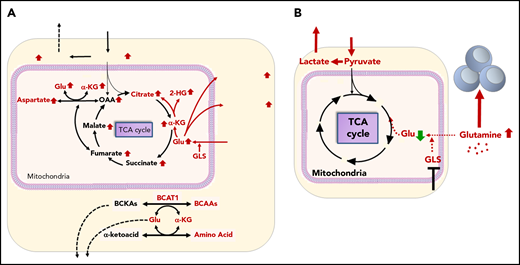
Gln metabolism in AML in tumor microenvironment. (A) Tumor cells avidly consume Gln to fuel biosynthesis and proliferation. GLS converts Gln to Glu, which is a necessary step for Gln to enter the TCA cycle. BCAT1, a cytosolic aminotransferase for BCAAs (leucine, isoleucine, and Val), is a critical enzyme that initiates the catabolism of BCAAs and results in the production of Glu. (B) Consumption of Gln by tumor cells deprives immune cells of this critical nutrient. GLS inhibitor blocks Gln consumption by tumor cells and elevates Glu in the tumor microenvironment supporting immune cell function.
Gln metabolism in AML in tumor microenvironment. (A) Tumor cells avidly consume Gln to fuel biosynthesis and proliferation. GLS converts Gln to Glu, which is a necessary step for Gln to enter the TCA cycle. BCAT1, a cytosolic aminotransferase for BCAAs (leucine, isoleucine, and Val), is a critical enzyme that initiates the catabolism of BCAAs and results in the production of Glu. (B) Consumption of Gln by tumor cells deprives immune cells of this critical nutrient. GLS inhibitor blocks Gln consumption by tumor cells and elevates Glu in the tumor microenvironment supporting immune cell function.
Recently, the ASNase and GLNase activities of 4 different isoforms of ASNase were investigated: E coli ASNase, ERWase, and native and mutant isoforms of Helicobacter pylori ASNase (HpA), the native wild-type (wt) HpA and the mutant developed mutant (dm) HpA.82 All 4 ASNase isoforms had similar ASNase activities, but ERWase exhibited the highest and dm HpA showed the lowest GLS activities. Of the 4, ERWase exhibited the highest cytotoxic activity on ALL samples, indicating an important role for the GLS activity of ASNase in the drug’s overall anticancer activity. Asn-dependent ALL cells showed no difference in sensitivity to any of the 4 tested isoforms.82
Notably, a direct correlation between obesity and the incidence of relapse in children with ALL has been reported. Parmentier et al82 and Ehsanipour et al83 reported the counteractive effects of BM adipocytes on leukemia cells after ASNase treatment. They demonstrated that the Gln-synthetase production by BM adipocytes was upregulated upon exposure to chemotherapy, although there was no change in plasma levels of Gln or Asn. Similarly, in xenograft models, adipocytes inhibited the cytotoxic activity of ASNase by releasing Gln into the leukemia microenvironment. Both native and pegylated forms of ASNase prolonged survival in nonobese mice with ALL; however, pegylated ASNase failed to prolong survival in their obese counterparts. Furthermore, although adipocytes secrete both Asn and Gln, Asn did not protect ALL cells from the cytotoxic effects of ASNase, and blocking Gln synthetase in adipocytes enhanced ASNase activity.83
Given the pivotal role of Gln in tumor cell survival, direct targeting of this pathway has been pursued through pharmacological GLS inhibitors. In preclinical studies, GLS inhibitor CB-839 decreased mitochondrial oxidative phosphorylation and inhibited AML cell growth.84 CB-839 further impaired antioxidant GSH production, which resulted in accumulation of mitochondrial ROS (mitoROS) followed by apoptosis induction in AML cells without cytotoxic effects on normal hematopoietic progenitors.85,86 The phase 1 clinical trial of CB-839 in relapsed/refractory AML demonstrated excellent tolerance but lack of objective responses when used as a single agent.87 A recent preliminary report of the ongoing combination trial of CB-839 and 5-azacitidine demonstrated rapid reduction of BM blasts in high-risk myelodysplastic syndrome patients.88 In preclinical studies with AML cell lines and patient samples harboring isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) mutations, CB-839 decreased production of oncometabolite 2-hydroxyglutarate, inducing differentiation,89 and the synergy between GLS inhibition and B-cell lymphoma 2 (BCL-2) inhibitor venetoclax has been reported.85 Gallipoli et al86 demonstrated the role of Gln on the metabolic adaptations supporting both mitochondrial function and cellular redox metabolism as resistance mechanisms of FLT3 internal tandem duplication (FLT3-ITD)–mutated AML to direct FLT3 kinase inhibitors. FLT3 inhibitors quizartinib has been shown to inhibit glucose uptake and glycolysis, rendering cells dependent on Gln metabolism; in turn, the combination of CB-839 with AC220 synergistically depleted GSH, induced mitoROS, and caused apoptotic cell death in both in vitro and in vivo mouse models.86,90 These studies indicate that GLS inhibition in combination with other targeted agents including BCL-2 and FLT3 tyrosine kinase inhibitors may offer therapeutic strategies targeting metabolism and survival in AML. Currently, the combination of CB839 and hypomethylating agent azacitidine is ongoing in high-risk myelodysplastic syndrome patients.88
Recently, the oncogenic function of branched-chain aminotransferase 1 (BCAT1), a cytosolic aminotransferase for branched-chain amino acids (BCAAs; leucine, isoleucine, and Val), was described as a critical enzyme for α-ketoglutarate (αKG) homeostasis related to leukemia metabolism.91,92 BCAT1 transfers α-amino groups from BCAAs to αKG, which results in the production of Glu (Figure 4). BCAT1 has been shown to be enriched and activated in LSCs in human AML and blast-crisis chronic myeloid leukemia, linking BCAA metabolism to epigenomic and posttranslational hypoxia-inducible factor 1α (HIF1α) regulation via αKG-dependent dioxygenases.93 Raffel et al demonstrated that knockdown of BCAT1 caused accumulation of αKG and HIF1α protein degradation, which translated into a growth-and-survival defect and abrogated leukemia-initiating potential. In turn, overexpression of BCAT1 decreased intracellular αKG levels and caused DNA hypermethylation through altered TET activity, similar to the phenotype exhibited by leukemia cells harboring mutant IDH.93 These findings suggest that αKG is a naturally occurring metabolite with tumor-suppressive potential, and suggest the BCAA-BCAT1-αKG pathway as a therapeutic target to compromise LSC function.
Cysteine
Cys is a semiessential amino acid. Previous studies highlighted the elevated intracellular levels of ROS in cancer cells, a result of genetic and microenvironment alterations and high metabolism rate.94 Those high levels of ROS contribute directly to oxidative stress. To counterbalance high levels of ROS, tumor cells enhance the production of reducing equivalents in part through maintenance of the reduced form of GSH.94,95 Because Cys is 1 of the building blocks of GSH, elevated production of GSH may exhaust endogenous sources of Cys.96 Subsequently, to survive and proliferate, cancer cells accelerate the uptake of extracellular cystine (CSSC; Cys disulfide) via the CSSC/Glu antiporter (xCT transporter).96,97
Cysteinase, which decreases Cys levels in serum and depletes extracellular Cys, is a potential cancer therapeutic tool.98 To address pharmacokinetic limitations including short half-life, Cramer et al98 engineered cysteinase conjugated to PEG. An in vivo study in cynomolgus monkeys showed that the engineered cysteinase consumed both CSSC and Cys with a high kinetic rate and impeded survival of HMVP2 prostate cancer cells. Cysteinase treatment caused an increase in intracellular ROS accumulation and increased autophagy by elevating 5′ adenosine monophosphate-activated protein kinase (AMPK) phosphorylation and decreasing mammalian target of rapamycin (mTOR) phosphorylation at Ser 2448.98,-100 In a murine model, intraperitoneal administration of cysteinase decreased. CSSC and Cys levels in serum were almost completely eliminated, but the levels recovered to preinjection concentrations after 4 days and 2 days, respectively. No side effects such as weight loss or organ anomalies were observed in either model. Cysteinase treatment significantly inhibited proliferation of the DU145 (P < .0001 at 100 mg/kg) and PC3 (P < .0001 at 50 and 100 mg/kg) prostate cancer cell lines in xenograft murine models.98
Zhang et al96 demonstrated that, whereas primary chronic lymphoid leukemia (CLL) cells exhibited a limited capacity to transport CSSC into the cells due to low expression of xCT, the cells effectively imported Cys produced by BM stromal cells, which expressed high levels of xCT, imported CSSC, converted it to Cys, and exported Cys into the BM microenvironment. The authors also highlighted the reliance of CLL cells on extracellular Cys for synthesizing GSH, which enhanced CLL cell survival and protected CLL cells from drug-induced cytotoxicity.96 That stroma-leukemia interaction represents 1 of the key amino acid–metabolic pathways for effectively targeting residual leukemia cells in the BM microenvironment (Figure 1).
Very recently, it has been reported that LSCs are auxotrophic to Cys to sustain energy metabolism and that targeting this axis with a cysteine-degrading enzyme may represent an effective therapeutic strategy.101 Cys depletion led to impaired GSH synthesis, decreased glutathionylation of succinate dehydrogenase A (a key component of electron transport chain complex II), and decreased oxidative phosphorylation, and, consequently, decreased production of adenosine triphosphate, leading to LSC death.101
To mimic the malignant BM microenvironment, Carmer et al98 cocultured splenocytes from mice bearing the aggressive TLC1-Tg:p53−/− CLL, which develop disease resembling human CLL, with BM stromal cells. They observed that cysteinase effectively decreased the viability of those splenocytes. Furthermore, TLC1-Tg:p53−/− CLL-bearing mice treated with cysteinase monotherapy showed a significantly higher survival rate (median, 7 months) than the untreated mice (median, 3.5 months; P < .0001). Cysteinase as monotherapy or combined with fludarabine, tested on CLL with 17p deletions (which is more aggressive and resistant to standard of drugs than CLL without these deletions),102 were both shown to be effective in killing the CLL cells in coculture with stromal cells.98
Studies of cysteine have prompted the need for attention to the importance of media usage in the context of in vitro metabolomics studies. Cantor et al103 demonstrated that traditional standard culture media and mouse plasma poorly reflect the metabolite composition of human plasma. On the other hand, culture media with polar metabolite concentrations comparable to those of human plasma yielded physiologically relevant alterations of the cellular redox state, glucose utilization, and overall metabolome.103 Further physiological relevance with respect to discovery of unforeseen metabolic wiring and novel metabolite-drug interactions can be achieved through the incorporation of coculture with stromal cells to properly model the hematological microenvironment.
Immune tumor microenvironment
The tumor microenvironment mediates resistance to amino acid–depletion therapies. Recent insights into the immunological component of the tumor microenvironment highlight the functional interplay between tumor cells, amino acids, and immune cells.104
In probing the mechanism by which patients with AML manifest lymphopenia and pancytopenia, Mussai et al showed that AML blasts and myeloid-derived suppressor cells (MDSCs) exhibit similar capacity to block the proliferation of T cells and human hematopoietic progenitor cells.105 They further demonstrated that MDSCs express ARGase I, and AML blasts secrete ARGase II, both of which induce the specific M-2 phenotype in the surrounding monocytes to suppress immunity. Those findings are consistent with the clinical symptoms of lymphopenia and pancytopenia in AML patients and confirm the immunosuppressive activity of AML blasts in blood and BM.105 (Figure 2) Furthermore, Steggerda et al highlighted the suppressive mechanism of granulocytes, myeloid cells that reside in the peripheral blood and aggregate to the site of infection or injury. Once activated, granulocytes release ARGase I from the cytosol to the extracellular microenvironment. ARGase I consumes the Arg required for immune cell proliferation by converting it to ornithine, thereby impeding proliferation of T cells and natural killer cells. The authors found that a potent and selective ARGase I inhibitor, CB-1158, inhibits Arg depletion in culture medium. Notably, the drug restored proliferation of MDSCs and granulocytes purified from patients with lung or head and neck cancer.106
GLS inhibitors, which block Gln consumption, and ARGase inhibitors, which block Arg consumption, increase availability of their respective target nutrients for immune cells (Figure 4).107 Ongoing clinical trials are exploring these inhibitors as monotherapy and in combination with anti-PD1 blockade.108 The results summarized here, however, provide a cautionary note for the development of therapies that deplete Arg because Arg can both positively modulate AML growth and negatively modulate an immunosuppressive microenvironment. The question of how AML cells tolerate Arg depletion mediated by self-produced ARGase II in their microenvironment remains unknown. It is possible that AML cells maintain a delicate balance between reserved intracellular Arg and secreted Arg and/or activate alternative metabolic pathways to survive when Arg is depleted. To mitigate negative impact on the AML microenvironment and effectively eradicate AML cells, adjunct approaches such as combinations of immune therapies and amino acid–targeted therapies should be evaluated.
Conclusions
Hematological malignancies frequently develop dependency on specific amino acids for their survival, and insufficient production of these amino acids creates a metabolic vulnerability and therapeutic opportunity.109 Amino acid–targeted enzymes such as ASNase, the first enzyme approved by the US Food and Drug Administration for the treatment of ALL, have shown promising results.12 For 5 decades, ASNase has been classified as a chemotherapy, but as highlighted in this review, ASNase and other amino acid–targeting enzymes fulfill the major requirements of targeted therapies: targeting a specific molecule that is upregulated or specific to cancer cells over normal cells. Therefore, we encourage the oncology community to reclassify ASNase and similar amino acid–targeted enzymes as targeted therapies.
Antimetabolites that interrupt amino acid synthesis have also been developed and are undergoing clinical trials as cancer therapeutics.110,-112 However, the lack of antimetabolite specificity often leads to undesirable side effects. Moreover, cancer cells can develop resistance due to upregulation of endogenous enzymes, collateral pathways, and the microenvironment when used as single agents.81 Therefore, drug combinations are being studied extensively to optimize amino acid–targeted enzyme therapy. In fact, combination of the BCL-2 inhibitor venetoclax with the hypomethylating agent azacitidine acts to inhibit amino acid uptake and catabolism, and the combination has been shown to be effective against LSCs, which depend on amino acid metabolism for survival.109 These findings provide a molecular mechanism for targeting LSCs and indicate that clinically relevant eradication of LSCs can be achieved with drugs that target LSC metabolic vulnerabilities.109 On the other hand, amino acid–modulating therapies that enhance the supply of amino acids to tumor-infiltrating lymphocytes may function as adjunct approaches in immune therapies.
Acknowledgments
The authors thank Sanaz Ghotbaldini for assistance with manuscript preparation, and Kathryn Hale, Department of Scientific Publications, The University of Texas Anderson Cancer Center, for manuscript review.
This work was supported in part by the National Institutes of Health, National Cancer Institute (R01 CA206210-01) (M.K.).
Authorship
Contribution: All authors have contributed to the preparation of the manuscript and reviewed/approved it in its final form.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marina Konopleva, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Unit 428, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: mkonople@mdanderson.org.
