

Key Points
Cfz decreases left ventricular function in mice through increased PP2A activity and inhibition of AMPKα/autophagy regulatory axes.
Met preserves left ventricular function in mice by restoring AMPKα activation; thus, it emerges as a prophylactic therapy.

Abstract
Carfilzomib (Cfz), an irreversible proteasome inhibitor licensed for relapsed/refractory myeloma, is associated with cardiotoxicity in humans. We sought to establish the optimal protocol of Cfz-induced cardiac dysfunction, to investigate the underlying molecular-signaling and, based on the findings, to evaluate the cardioprotective potency of metformin (Met). Mice were randomized into protocols 1 and 2 (control and Cfz for 1 and 2 consecutive days, respectively); protocols 3 and 4 (control and alternate doses of Cfz for 6 and 14 days, respectively); protocols 5A and 5B (control and Cfz, intermittent doses on days 0, 1 [5A] and 0, 1, 7, and 8 [5B] for 13 days); protocols 6A and 6B (pharmacological intervention; control, Cfz, Cfz+Met and Met for 2 and 6 days, respectively); and protocol 7 (bortezomib). Cfz was administered at 8 mg/kg (IP) and Met at 140 mg/kg (per os). Cfz resulted in significant reduction of proteasomal activity in heart and peripheral blood mononuclear cells in all protocols except protocols 5A and 5B. Echocardiography demonstrated that Cfz led to a significant fractional shortening (FS) depression in protocols 2 and 3, a borderline dysfunction in protocols 1 and 4, and had no detrimental effect on protocols 5A and 5B. Molecular analysis revealed that Cfz inhibited AMPKα/mTORC1 pathways derived from increased PP2A activity in protocol 2, whereas it additionally inhibited phosphatidylinositol 3-kinase/Akt/endothelial nitric oxide synthase pathway in protocol 3. Coadministration of Met prevented Cfz-induced FS reduction and restored AMPKα phosphorylation and autophagic signaling. Conclusively, Cfz decreased left ventricular function through increased PP2A activity and inhibition of AMPKα and its downstream autophagic targets, whereas Met represents a novel promising intervention against Cfz-induced cardiotoxicity.
Introduction
Multiple myeloma (MM) is a plasma cell malignancy1,2 in which proteasome inhibitors (PI) are widely used for its treatment.3 Carfilzomib (Cfz) is an irreversible PI licensed for the treatment of relapsed/refractory MM.4 The Carfilzomib and Dexamethasone Versus Bortezomib and Dexamethasone for Patients With Relapsed or Refractory Multiple Myeloma study showed that Cfz significantly improved survival of relapsed/refractory MM patients compared with treatment with the reversible PI bortezomib (Btz).5 However, Cfz (contrary to Btz) has been associated with severe cardiac adverse effects.1,6 MM is a disease of the elderly,7 and approximately two-thirds of patients had a concomitant coronary heart disease or other cardiovascular disorder before Cfz treatment.8 Additionally, patients with coexisting cardiovascular diseases and other comorbidities have a higher prevalence of developing cardiac dysfunction during Cfz treatment.9 Thus, thorough patient evaluation and risk assessment before initiation of therapy with Cfz is crucial.10,11
Notably, there are data lacking on current preventive strategies for Cfz-induced cardiotoxicity.10-15 Thus, there is an imperative need for molecular-based research on novel pharmacological interventions.
The aim of this study was (1) to establish an in vivo protocol of Cfz treatment with clinically relevant doses and to monitor cardiac function by echocardiography, (2) to elucidate the underlying mechanisms and to identify potential molecular targets, and (3) based on our findings, to investigate the ability of metformin (Met) to prevent Cfz-induced cardiac damage.
Materials and methods
Additional method details are described in the supplemental Data, available on the Blood Web site.
Animals
A total of 187 male C57BL/6 mice, 12 weeks old, weighting ∼25 g, were housed under normal conditions and had free access to food and water (ad libitum). Mice were monitored daily and body weight variations were recorded. All animals received proper care in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals, prepared by the Academy of Sciences and published by the National Institutes of Health. All animal procedures were approved by the Ethical Committee of the Prefecture of Athens and were implemented at the Biomedical Research Foundation of Academy of Athens (protocol no. 1757, Athens, 04/04/2017; 1763, Athens, 04/04/2017).
Experimental protocols
All experimental protocols are illustrated in supplemental Figure 1. Cfz was administered at 8 mg/kg (IP). In all protocols, animals were anesthetized and underwent echocardiographic evaluation. Subsequently, animals were euthanized with ketamine overdose (ketamine 300 mg/kg) to obtain myocardial and blood samples for molecular analysis. No lethality was observed in control or Cfz-treated groups.
Protocol 1 (1 day).
Eleven mice (control, n = 5; Cfz, n = 6) received 1 dose normal saline (NS) or Cfz on day 0 (1 dose in total).
Protocol 2 (2-day protocol).
Fourteen mice (control, n = 5; Cfz, n = 9) received NS or Cfz on days 0 and 1 (2 doses in total).
Protocol 3 (alternate-acute).
Twenty-one mice (control, n = 7; Cfz, n = 14) received NS or Cfz every 48 hours for 6 days (4 doses in total).
Protocol 4 (alternate subchronic).
Nineteen mice (control, n = 8; Cfz, n = 11) received NS or Cfz on day 0 and every 48 hours for 14 days (8 doses in total).
Protocol 5A and 5B (intermittent, acute, and subchronic).
Twenty eight mice (control, n = 14; Cfz, n = 14) received NS or Cfz on days 0, 1 (protocol 5A) and 0, 1, 7, 8 (protocol 5B) and undergoing washout periods between days 2 and 6 and 9 and 13.
Protocol 6A (pharmacological intervention-2 dose Met).
Thirty mice (control, n = 8; Cfz, n = 9; Cfz+Met, n = 9; Met, n = 4) received Cfz and Met (140 mg/kg, per os) on days 0 and 1 (2 doses in total). Metformin was isolated from Glucophage and its purity was verified by nuclear magnetic resonance analysis (supplemental Figure 2).
Protocol 6B (pharmacological intervention-6D Met).
Forty-four mice (control, n = 13; Cfz, n = 14; Cfz+Met, n = 11; Met, n = 6) received Cfz every 48 hours and Met (140 mg/kg, per os) daily for 6 days. Fasting glucose levels were monitored on days 0 and 6.
Protocol 7 (Btz).
Twenty mice (control, n = 6; Btz, n = 14) received NS or Btz (1 mg/kg, IP) respectively, on day 0 and every 48 hours for 6 days (4 doses in total). Because of increased mortality presented in the Btz group, 8 mice completed the study.
Biometric data on body weight alterations of protocols 3, 4, 5A, 5B, and 6B are presented in supplemental Figure 3 and show no significant changes between groups.
Echocardiography
At the beginning and at the end of these protocols, mice were anesthetized (ketamine, 100 mg/kg IP) and underwent echocardiographic assessment using an ultrasound system (Vivid 7; GE Healthcare) with a 13-MHz linear transducer, as previously described.16 Heart rate, left ventricular end-diastole (LVEDD) and left ventricular end-systole diameter (LVESD), left ventricular posterior wall thickness at diastole (LVPWd) and at systole (LVPWs), fractional shortening (FS; FS% = (LVEDD − LVESD) / LVEDD × 100%) and left ventricular radius to left ventricular posterior wall thickness ratio were calculated.
Histology
Tissue samples for histopathological evaluation were fixed in 4% buffered formalin solution for 24 hours and embedded in paraffin waxes. Three-micrometer-thick sections were obtained from each sample and routinely counterstained with hematoxylin and eosin for histological examination as described in the supplemental Methods.
Measurement of proteasome peptidase activity
To validate the selected Cfz dose and evaluate proteasome inhibition-related effects in all study protocols, the chymotrypsin-like (CT-L, LLVY/β5; rate limiting for protein breakdown) proteasomal activity was measured in heart tissue and peripheral blood mononuclear cells (PBMC) using a fluorogenic peptide method as described in supplemental Methods.17 Analyzed samples were pools from 3 to 6 animals/protocol.
Immunoblotting and PP2A activity measurement
Western blot analysis in myocardial tissue samples was performed as described previously.18 The activity of PP2A was measured using a PP2A immunoprecipitation phosphatase assay kit (Merck S.A. Hellas), per the manufacturer’s instructions.19 A detailed description is reported in supplemental Methods.
Statistics
Statistical analysis was performed using the GraphPad Prism 6 software; results were plotted in graphs as mean ± standard error of the mean values. Data were analyzed using 1-way analysis of variance (comparing the means among groups and using Tukey as a post hoc test for multiple comparisons) or 2-sided t test as required. Echocardiography data were analyzed using 1-way analysis of variance (comparing the means among groups and using Bonferroni as a post hoc test for multiple comparisons) or 2-sided t test (paired or unpaired) as required. P < .05 was considered statistically significant (*P < .05, **P < .01, ***P < .001).
Results
Mice treatment with Cfz suppressed proteasome activity
Following treatment of 1 or 2 consecutive days (protocols 1 and 2) and on alternate days (protocols 3 and 4), Cfz resulted in significant reduction (vs control) of the assayed CT-L proteasomal peptidase activity in both the myocardial tissue (Figure 1A; Figure 2A; supplemental Figure 4A) and PBMCs (Figure 1A; Figure 2A; supplemental Figure 4A). In protocols 5A and 5B (intermittent, acute, and subchronic), Cfz did not reduce the CT-L proteasomal activity in myocardium (Figure 1A; Figure 2A), whereas in PBMCs, it only reduced CT-L proteasomal peptidase activity in protocol 5A (Figure 1B).
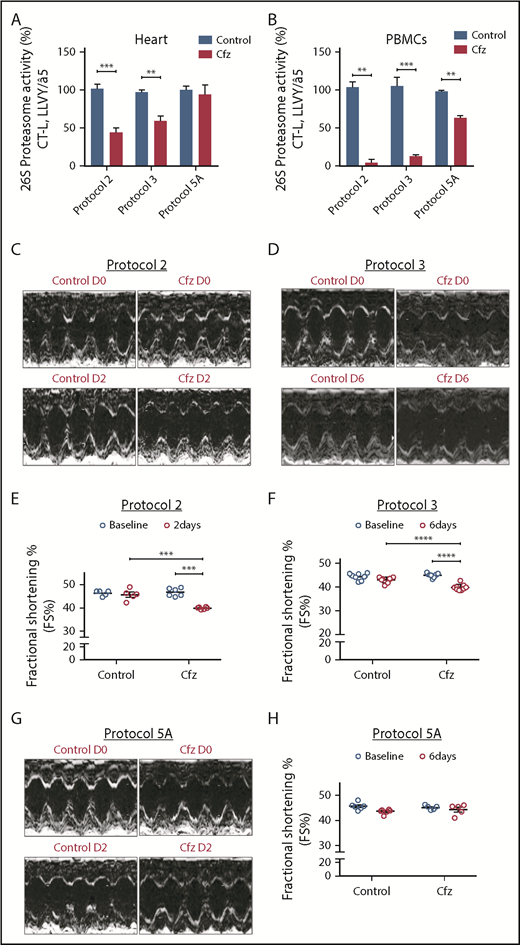
Proteasomal activity and cardiac function in acute Cfz protocols. (A) Proteasomal activity in myocardial tissue in experimental protocols 2, 3, and 5A. (B) Proteasomal activity in PBMCs in experimental protocols 2, 3, and 5A. (C) Representative echocardiographic M-mode images of protocol 2 at baseline and end point. (D) Representative echocardiographic M-mode images of protocol 3 at baseline and end point. (E) FS% graph of protocol 2 (circles, individual values). (F) FS% graph of protocol 3 (circles, individual values). (G) Representative echocardiographic M-mode images of protocol 5A at baseline and end point. (H) FS% graph of protocol 5A (circles, individual values). **P < .01, ***P = .001, ****P < .001; n = 3-6 per group.
Proteasomal activity and cardiac function in acute Cfz protocols. (A) Proteasomal activity in myocardial tissue in experimental protocols 2, 3, and 5A. (B) Proteasomal activity in PBMCs in experimental protocols 2, 3, and 5A. (C) Representative echocardiographic M-mode images of protocol 2 at baseline and end point. (D) Representative echocardiographic M-mode images of protocol 3 at baseline and end point. (E) FS% graph of protocol 2 (circles, individual values). (F) FS% graph of protocol 3 (circles, individual values). (G) Representative echocardiographic M-mode images of protocol 5A at baseline and end point. (H) FS% graph of protocol 5A (circles, individual values). **P < .01, ***P = .001, ****P < .001; n = 3-6 per group.
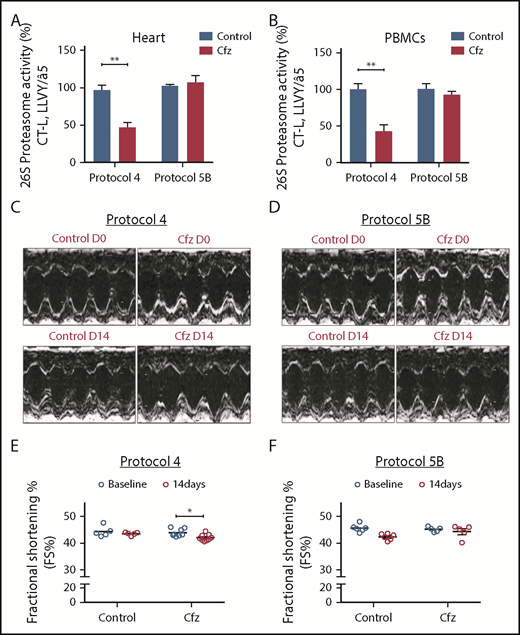
Proteasomal activity and cardiac function in subchronic Cfz protocols. (A) Proteasomal activity in myocardial tissue in experimental protocols 4 and 5B. (B) Proteasomal activity in PBMCs in experimental protocols 4 and 5B. (C) Representative echocardiographic M-mode images of protocol 4 at baseline and end point. (D) Representative echocardiographic M-mode images of protocol 5B at baseline and end point. (E) FS% graph of protocol 4 (circles, individual values). (F) FS% graph of protocol 5B (circles, individual values). *P < .05, **P < .01.
Proteasomal activity and cardiac function in subchronic Cfz protocols. (A) Proteasomal activity in myocardial tissue in experimental protocols 4 and 5B. (B) Proteasomal activity in PBMCs in experimental protocols 4 and 5B. (C) Representative echocardiographic M-mode images of protocol 4 at baseline and end point. (D) Representative echocardiographic M-mode images of protocol 5B at baseline and end point. (E) FS% graph of protocol 4 (circles, individual values). (F) FS% graph of protocol 5B (circles, individual values). *P < .05, **P < .01.
Acute Cfz administration reduced FS without major histological changes
Echocardiography analysis in the Cfz group (protocol 1) demonstrated a statistically significant, albeit moderate, reduction of FS compared with control (supplemental Table 1; supplemental Figure 4B-C). In protocols 2 and 3, a significant reduction in FS% of Cfz-treated mice vs controls was observed (Figure 1C-F; Tables 1 and 2; supplemental Tables 2 and 3). The Cfz group in protocol 3 showed a significantly reduced (compared with control) thickness in LVPWd and LVPWs. Interestingly, when Cfz was administered for 2 consecutive days (protocol 5A) and the examination was performed on day 6 (4 days of washout period), Cfz did not result in cardiac dysfunction (Figure 1G-H; supplemental Table 5). Histology assessment in heart tissues of mice enrolled in protocol 3 showed no significant changes between control and Cfz groups for nuclear irregularity, vessel congestion, and edema; relative score values ranged from absent to mild (supplemental Figure 5A-B).
Echocardiography results of protocol 2
| Protocol 2, day 2 | Control | Cfz |
|---|---|---|
| n = 5 | n = 6 | |
| HR | 651.46 ± 16.02 | 648.38 ± 23.15 |
| LVEDD, mm | 3.06 ± 0.07 | 3.28 ± 0.11 |
| LVESD, mm | 1.66 ± 0.06 | 1.97 ± 0.07* |
| LVPWd, mm | 0.75 ± 0.01 | 0.73 ± 0.01 |
| LVPWs, mm | 1.27 ± 0.01 | 1.24 ± 0.01 |
| FS, % | 45.67 ± 1.07 | 39.88 ± 0.20*** |
| r/h | 2.07 ± 0.06 | 2.21 ± 0.09 |
| Protocol 2, day 2 | Control | Cfz |
|---|---|---|
| n = 5 | n = 6 | |
| HR | 651.46 ± 16.02 | 648.38 ± 23.15 |
| LVEDD, mm | 3.06 ± 0.07 | 3.28 ± 0.11 |
| LVESD, mm | 1.66 ± 0.06 | 1.97 ± 0.07* |
| LVPWd, mm | 0.75 ± 0.01 | 0.73 ± 0.01 |
| LVPWs, mm | 1.27 ± 0.01 | 1.24 ± 0.01 |
| FS, % | 45.67 ± 1.07 | 39.88 ± 0.20*** |
| r/h | 2.07 ± 0.06 | 2.21 ± 0.09 |
Echocardiographic assessment of cardiac function in Cfz-treated mice indicated a moderate but significant LV dilation and FS% reduction vs control group at day 2 (*P < .05, **P < .01, ***P < .001).
HR, heart rate (beats/min); r/h: left ventricular radius to left ventricular posterior wall thickness ratio.
Echocardiography results of protocol 3
| Protocol 3, day 6 | Control | Cfz |
|---|---|---|
| n = 7 | n = 8 | |
| HR | 636.28 ± 14.66 | 585.92 ± 10.58* |
| LVEDD, mm | 3.03 ± 0.07 | 3.08 ± 0.09 |
| LVESD, mm | 1.73 ± 0.04 | 1.86 ± 0.06 |
| LVPWd, mm | 0.76 ± 0.01 | 0.69 ± 0.01** |
| LVPWs, mm | 1.24 ± 0.02 | 1.17 ± 0.01** |
| FS, % | 43.03 ± 0.50 | 39.87 ± 0.47*** |
| r/h | 2.01 ± 0.05 | 2.21 ± 0.09 |
| Protocol 3, day 6 | Control | Cfz |
|---|---|---|
| n = 7 | n = 8 | |
| HR | 636.28 ± 14.66 | 585.92 ± 10.58* |
| LVEDD, mm | 3.03 ± 0.07 | 3.08 ± 0.09 |
| LVESD, mm | 1.73 ± 0.04 | 1.86 ± 0.06 |
| LVPWd, mm | 0.76 ± 0.01 | 0.69 ± 0.01** |
| LVPWs, mm | 1.24 ± 0.02 | 1.17 ± 0.01** |
| FS, % | 43.03 ± 0.50 | 39.87 ± 0.47*** |
| r/h | 2.01 ± 0.05 | 2.21 ± 0.09 |
Echocardiographic assessment of cardiac function in mice demonstrates significant reduction in FS% in Cfz vs control group (P < .001) at day 6 (*P < .05, **P < .01, ***P < .001).
Subchronic Cfz administration demonstrated LV dilation
Subchronic administration of Cfz (protocol 4) resulted in LV dilation on day 14 compared with control. However, overall cardiac function was not significantly affected as presented by the borderline decreased FS (Figure 2C,E; supplemental Table 4). Intermittent Cfz dosing (protocol 5B) did not result in cardiac dysfunction (Figure 2D,F; supplemental Table 5). Histological evaluation demonstrated no major differences for the subchronic Cfz administration (supplemental Figure 5A-B).
Cfz administration induced cardiotoxicity mainly through inhibition of AMPKα phosphorylation and autophagy-related proteins
Based on the echocardiography results, we investigated the molecular mechanisms of Cfz-induced cardiotoxicity in protocols 1, 2, and 3. One injection of Cfz tended to reduce AMPKα and Raptor phosphorylation as well as expression of LC3-II; yet, these effects did not reach statistical significance (supplemental Figure 4D-E). Two consecutive injections of Cfz resulted in significantly decreased AMPKα phosphorylation (Figure 3A-B). By investigating the downstream targets of AMPKα, namely Raptor, mTOR, and LC3-II, we observed that Cfz decreased Raptor phosphorylation, did not change mTOR phosphorylation, and reduced the expression of LC3-II. We did not observe any significant differences in phosphatidylinositol 3-kinase (PI3K), Akt, or endothelial nitric oxide synthase (eNOS) phosphorylation (Figure 3A-B).
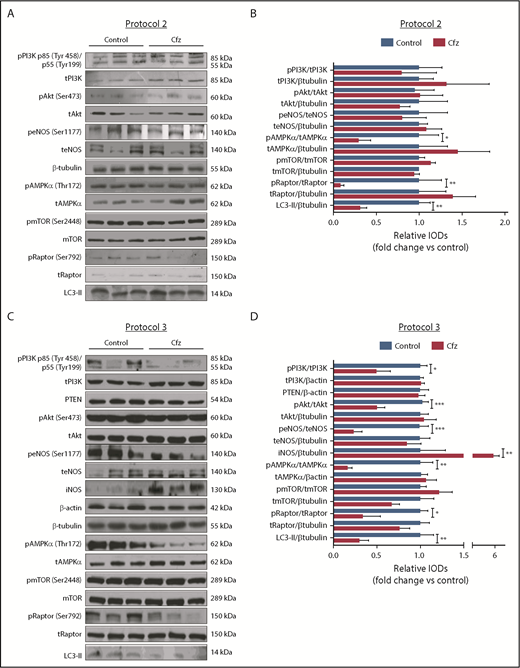
Cfz-induced cardiotoxicity mechanism: acute Cfz administration induced cardiotoxicity primarily through inhibition of AMPKα-mediated autophagy and PI3K/Akt/eNOS axis. (A) Representative western blots of protocol 2. (B) Representative column graphs and densitometry analysis of protocol 2: PI3K p85(Tyr458)/p55(Tyr199)/tPI3K, tPI3K/β-tubulin, pAkt(Ser473)/tAkt, tAkt/β-tubulin, peNOS(Ser1177)/teNOS, t-eNOS/β-tubulin, pAMPKα(Thr172)/tAMPKα, tAMPKα/β-tubulin, pmTOR(Ser2448)/tmTOR, tmTOR/β-tubulin, pRaptor(Ser792)/tRaptor, tRaptor/β-tubulin, LC3-II/ β-tubulin. (C) Representative western blots of protocol 3. (D) Representative column graphs and densitometry analysis of protocol 3: PI3K p85(Tyr458)/p55(Tyr199)/tPI3K, tPI3K/β-actin, PTEN/β-actin, pAkt(Ser473)/tAkt, tAkt/β-tubulin, peNOS(Ser1177)/teNOS, t-eNOS/β-tubulin, iNOS/β-tubulin, pAMPKα(Thr172)/tAMPKα, tAMPKα/β-actin, pmTOR(Ser2448)/tmTOR, tmTOR/β-tubulin, pRaptor(Ser792)/tRaptor, tRaptor/β-tubulin, LC3-II/β-tubulin. *P < .05, *P < .01, ***P < .001 n = 4-9 per group. In each protocol, 1 representative western blot image for the loading controls (ie, β-tubulin and/or β-actin) is presented for ergonomic reasons. Not all proteins ran on the same sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Western blots show 3 representative animals from each experimental group, 1 in each lane. IOD, integral optical density.
Cfz-induced cardiotoxicity mechanism: acute Cfz administration induced cardiotoxicity primarily through inhibition of AMPKα-mediated autophagy and PI3K/Akt/eNOS axis. (A) Representative western blots of protocol 2. (B) Representative column graphs and densitometry analysis of protocol 2: PI3K p85(Tyr458)/p55(Tyr199)/tPI3K, tPI3K/β-tubulin, pAkt(Ser473)/tAkt, tAkt/β-tubulin, peNOS(Ser1177)/teNOS, t-eNOS/β-tubulin, pAMPKα(Thr172)/tAMPKα, tAMPKα/β-tubulin, pmTOR(Ser2448)/tmTOR, tmTOR/β-tubulin, pRaptor(Ser792)/tRaptor, tRaptor/β-tubulin, LC3-II/ β-tubulin. (C) Representative western blots of protocol 3. (D) Representative column graphs and densitometry analysis of protocol 3: PI3K p85(Tyr458)/p55(Tyr199)/tPI3K, tPI3K/β-actin, PTEN/β-actin, pAkt(Ser473)/tAkt, tAkt/β-tubulin, peNOS(Ser1177)/teNOS, t-eNOS/β-tubulin, iNOS/β-tubulin, pAMPKα(Thr172)/tAMPKα, tAMPKα/β-actin, pmTOR(Ser2448)/tmTOR, tmTOR/β-tubulin, pRaptor(Ser792)/tRaptor, tRaptor/β-tubulin, LC3-II/β-tubulin. *P < .05, *P < .01, ***P < .001 n = 4-9 per group. In each protocol, 1 representative western blot image for the loading controls (ie, β-tubulin and/or β-actin) is presented for ergonomic reasons. Not all proteins ran on the same sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Western blots show 3 representative animals from each experimental group, 1 in each lane. IOD, integral optical density.
In protocol 3, Cfz decreased AMPKα phosphorylation and its downstream targets Raptor and LC3-II, whereas mTOR phosphorylation remained unchanged (Figure 3C-D). However, this dosage protocol resulted in reduction of PI3K, Akt, and eNOS phosphorylation without changes in PTEN. Because we observed a decrease in eNOS phosphorylation, we evaluated the effects of Cfz on iNOS expression and noticed a significant increase compared with the control group (Figure 3C-D).
Metformin prevented Cfz-induced FS% reduction
Given our previously findings, we coadministered Met (an AMPKα activator20 ) along with Cfz treatment to reactivate the AMPKα pathway. The coadministration of Met either for 2 or 6 days did not interfere with the proteasomal inhibitory activity of Cfz in both the myocardium and PBMCs (Figure 4A-B). Under these conditions, Met resulted in abrogation of the Cfz-induced defects, which was evident by the elevation of FS% in protocols 6A and 6B (Figures 4C-F; Tables 3 and 4; supplemental Tables 6 and 7). Met did not reduce fasting glucose levels (supplemental Figure 3D). In protocol 6B, the LVPWd and LVPWs thickness presented a borderline reduction in the Cfz+Met group compared with control, but remained within physiological limits. In comparison with day 0, the Cfz+Met group had no significant changes in the echocardiographic parameters within the same group (supplemental Table 7). Histological examination of heart tissues from protocol 6B demonstrated a mild to moderate increase in vascular congestion in Cfz+Met and Met groups in comparison with either the control or Cfz group (supplemental Figure 5A-B); we considered this a nonspecific finding that does not constitute a permanent lesion because we found no signs of hemorrhage or vascular obstruction. Vascular congestion observed in the Cfz+Met and Met groups was defined as hyperemia, a process that in this instance was not linked to inflammation or other clinical alterations.
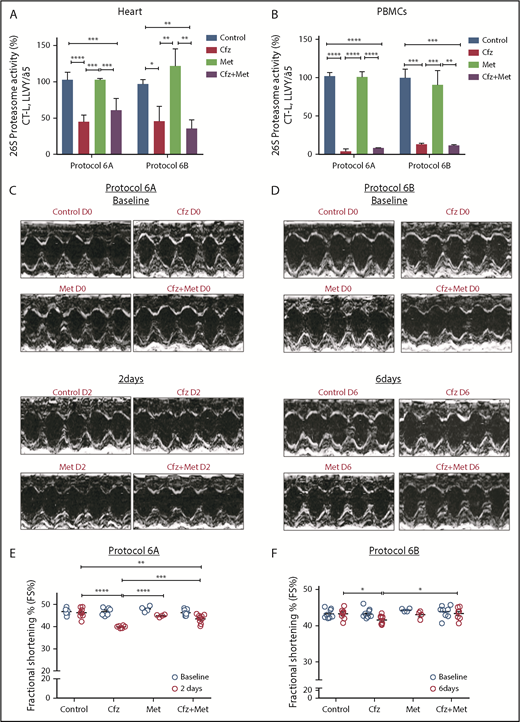
Coadministration of Met restored myocardial dysfunction without affecting Cfz proteasomal inhibitory activity. (A) Proteasomal activity in myocardial tissue in experimental protocols 6A and 6B. (B) Proteasomal activity in PBMCs in experimental protocols 6A and 6B. (C) Representative echocardiographic M-mode images of protocol 6A at baseline and end point. (D) Representative echocardiographic M-mode images of protocol 6B at baseline and end point. (E) FS% graph of protocol 6A (circles, individual values). (F) FS% graph of protocol 6B (circles, individual values). *P < .05, **P < .01, ***P = .001, ****P < .0001.
Coadministration of Met restored myocardial dysfunction without affecting Cfz proteasomal inhibitory activity. (A) Proteasomal activity in myocardial tissue in experimental protocols 6A and 6B. (B) Proteasomal activity in PBMCs in experimental protocols 6A and 6B. (C) Representative echocardiographic M-mode images of protocol 6A at baseline and end point. (D) Representative echocardiographic M-mode images of protocol 6B at baseline and end point. (E) FS% graph of protocol 6A (circles, individual values). (F) FS% graph of protocol 6B (circles, individual values). *P < .05, **P < .01, ***P = .001, ****P < .0001.
Echocardiography at pharmacological intervention protocol 6A
| Protocol 6A, day 2 | Control | Cfz | Cfz+Met | Met |
|---|---|---|---|---|
| n = 8 | n = 6 | n = 9 | n = 4 | |
| HR | 651.42 ± 10.07 | 648.38 ± 23.15 | 642.15 ± 12 | 647.16 ± 6.35 |
| LVEDD, mm | 3.04 ± 0.05* | 3.28 ± 0.11 | 3.18 ± 0.05 | 2.97 ± 0.11* |
| LVESD, mm | 1.61 ± 0.05****† | 1.97 ± 0.07 | 1.80 ± 0.05* | 1.63 ± 0.07*** |
| LVPWd, mm | 0.78 ± 0.01**†† | 0.73 ± 0.01 | 0.74 ± 0.01 | 0.78 ± 0.01**† |
| LVPWs, mm | 1.28 ± 0.01**†† | 1.24 ± 0.01 | 1.24 ± 0.01 | 1.29 ± 0.01**†† |
| FS, % | 46.20 ± 0.78****†† | 39.88 ± 0.20 | 43.49 ± 0.63*** | 44.94 ± 0.28**** |
| r/h | 1.98 ± 0.05* | 2.21 ± 0.09 | 2.15 ± 0.05 | 1.90 ± 0.08*† |
| Protocol 6A, day 2 | Control | Cfz | Cfz+Met | Met |
|---|---|---|---|---|
| n = 8 | n = 6 | n = 9 | n = 4 | |
| HR | 651.42 ± 10.07 | 648.38 ± 23.15 | 642.15 ± 12 | 647.16 ± 6.35 |
| LVEDD, mm | 3.04 ± 0.05* | 3.28 ± 0.11 | 3.18 ± 0.05 | 2.97 ± 0.11* |
| LVESD, mm | 1.61 ± 0.05****† | 1.97 ± 0.07 | 1.80 ± 0.05* | 1.63 ± 0.07*** |
| LVPWd, mm | 0.78 ± 0.01**†† | 0.73 ± 0.01 | 0.74 ± 0.01 | 0.78 ± 0.01**† |
| LVPWs, mm | 1.28 ± 0.01**†† | 1.24 ± 0.01 | 1.24 ± 0.01 | 1.29 ± 0.01**†† |
| FS, % | 46.20 ± 0.78****†† | 39.88 ± 0.20 | 43.49 ± 0.63*** | 44.94 ± 0.28**** |
| r/h | 1.98 ± 0.05* | 2.21 ± 0.09 | 2.15 ± 0.05 | 1.90 ± 0.08*† |
Echocardiographic assessment of cardiac function in protocol 6A demonstrated significant changes in all groups vs Cfz (*P < .05, ** P < .01, ***P < .001, ****P < .0001) and in the control and Met groups vs Cfz+Met group (†P < .05, ††P < .01), respectively.
Echocardiography at pharmacological intervention protocol 6B
| Protocol 6B, day 6 | Control | Cfz | Cfz+Met | Met |
|---|---|---|---|---|
| n = 8 | n = 8 | n = 8 | n = 4 | |
| HR | 630.04 ± 8.60 | 622.87 ± 9.66 | 609.75 ± 7.14 | 609.17 ± 15.98 |
| LVEDD, mm | 3.10 ± 0.07 | 3.07 ± 0.07 | 3.09 ± 0.05 | 3.09 ± 0.05 |
| LVESD, mm | 1.76 ± 0.05 | 1.79 ± 0.05 | 1.75 ± 0.04 | 1.76 ± 0.01 |
| LVPWd, mm | 0.77 ± 0.01 | 0.74 ± 0.01* | 0.74 ± 0.01* | 0.75 ± 0.01 |
| LVPWs, mm | 1.26 ± 0.01 | 1.24 ± 0.01 | 1.23 ± 0.01* | 1.23 ± 0.02 |
| FS, % | 43.24 ± 0.50 | 41.55 ± 0.43*† | 43.39 ± 0.56 | 43.03 ± 0.53 |
| r/h | 2.02 ± 0.06 | 2.06 ± 0.06 | 2.08 ± 0.06 | 2.04 ± 0.01 |
| Protocol 6B, day 6 | Control | Cfz | Cfz+Met | Met |
|---|---|---|---|---|
| n = 8 | n = 8 | n = 8 | n = 4 | |
| HR | 630.04 ± 8.60 | 622.87 ± 9.66 | 609.75 ± 7.14 | 609.17 ± 15.98 |
| LVEDD, mm | 3.10 ± 0.07 | 3.07 ± 0.07 | 3.09 ± 0.05 | 3.09 ± 0.05 |
| LVESD, mm | 1.76 ± 0.05 | 1.79 ± 0.05 | 1.75 ± 0.04 | 1.76 ± 0.01 |
| LVPWd, mm | 0.77 ± 0.01 | 0.74 ± 0.01* | 0.74 ± 0.01* | 0.75 ± 0.01 |
| LVPWs, mm | 1.26 ± 0.01 | 1.24 ± 0.01 | 1.23 ± 0.01* | 1.23 ± 0.02 |
| FS, % | 43.24 ± 0.50 | 41.55 ± 0.43*† | 43.39 ± 0.56 | 43.03 ± 0.53 |
| r/h | 2.02 ± 0.06 | 2.06 ± 0.06 | 2.08 ± 0.06 | 2.04 ± 0.01 |
Echocardiographic assessment of cardiac function in mice demonstrates significantly reduced FS% in Cfz vs control (*P < .05) and Cfz+Met groups (†P < .05) at day 6.
Met increased AMPKα phosphorylation and restored autophagy-related proteins
To confirm that AMPKα phosphorylation is one of the main mechanisms of Cfz-induced cardiotoxicity, we evaluated if the coadministration of Met increases AMPKα phosphorylation in both protocols. We observed that, indeed, treatment with Met for 2 days largely restored AMPKα phosphorylation and LC3-II expression (Figure 5A). Similarly, coadministration of Met with Cfz for 6 days successfully restored AMPKα phosphorylation in comparison with the Cfz group and increased Raptor phosphorylation and LC3-II expression vs Cfz; pmTOR had no significant changes compared with the control and Cfz groups (Figure 5C-D). The coadministration of Met did not change the levels of PTEN, or PI3K phosphorylation, whereas pAkt in the control and Cfz+Met groups was significantly increased vs the Cfz group (Figures 5C-D). In the Cfz+Met group, phosphorylation of eNOS (peNOS) and inducible nitric oxide synthase (iNOS) demonstrated a nonsignificant change compared with Cfz group (Figures 5C-D).
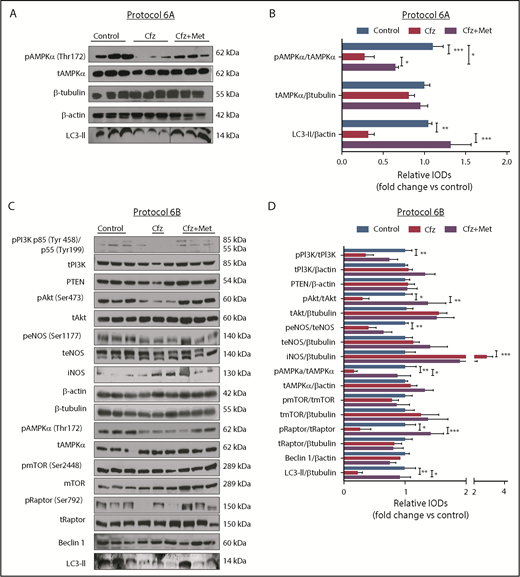
Met exerted cardioprotection mainly through restoring AMPKα phosphorylation and autophagy-related proteins. (A) Representative western blots of protocol 6A. (B) Representative column graphs and densitometry analysis of protocol 6A: pAMPKα(Thr172)/tAMPKα, tAMPKα/ β-tubulin, and LC3-II/ β-actin. (C) Representative western blots of protocol 6B. (D) Representative column graphs and densitometry analysis of protocol 3: PI3K p85(Tyr458)/p55(Tyr199)/tPI3K, tPI3K/β-actin, PTEN/β-actin, pAkt(Ser473)/tAkt, tAkt/β-tubulin, peNOS(Ser1177)/teNOS, teNOS/β-actin, iNOS/β-tubulin, pAMPKα(Thr172)/tAMPKα, tAMPKα/β-actin, pmTOR(Ser2448)/tmTOR, tmTOR/β-tubulin, pRaptor(Ser792)/tRaptor, tRaptor/β-tubulin, LC3-II/β-tubulin. *P < .05, *P < .01, ***P < .001; n = 4-9 per group for all the western blot. In each protocol, 1 representative western blot image for the loading controls (ie, β-tubulin and/or β-actin) is presented for ergonomic reasons. However not all proteins ran on the same sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Western Blots show 3 representative animals from each experimental group, 1 in each lane. Vertical line(s) have been inserted to indicate a repositioned gel lane.
Met exerted cardioprotection mainly through restoring AMPKα phosphorylation and autophagy-related proteins. (A) Representative western blots of protocol 6A. (B) Representative column graphs and densitometry analysis of protocol 6A: pAMPKα(Thr172)/tAMPKα, tAMPKα/ β-tubulin, and LC3-II/ β-actin. (C) Representative western blots of protocol 6B. (D) Representative column graphs and densitometry analysis of protocol 3: PI3K p85(Tyr458)/p55(Tyr199)/tPI3K, tPI3K/β-actin, PTEN/β-actin, pAkt(Ser473)/tAkt, tAkt/β-tubulin, peNOS(Ser1177)/teNOS, teNOS/β-actin, iNOS/β-tubulin, pAMPKα(Thr172)/tAMPKα, tAMPKα/β-actin, pmTOR(Ser2448)/tmTOR, tmTOR/β-tubulin, pRaptor(Ser792)/tRaptor, tRaptor/β-tubulin, LC3-II/β-tubulin. *P < .05, *P < .01, ***P < .001; n = 4-9 per group for all the western blot. In each protocol, 1 representative western blot image for the loading controls (ie, β-tubulin and/or β-actin) is presented for ergonomic reasons. However not all proteins ran on the same sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Western Blots show 3 representative animals from each experimental group, 1 in each lane. Vertical line(s) have been inserted to indicate a repositioned gel lane.
Bortezomib
To investigate if the acute cardiotoxicity by Cfz is a drug or class effect, we evaluated the effects of Btz on myocardium. We compared the effects of Btz and Cfz on myocardium in protocol 3 (alternate-acute) because it is clinically relevant for Btz. Administration of Btz (protocol 7) resulted in weight loss and increased mortality (supplemental Figure 6A-B) and significant proteasome inhibition in both myocardial tissue and PBMCs (supplemental Figure 6C), without exerting any cardiac functional damage (supplemental Table 8; supplemental Figure 6D). The Btz group demonstrated mild to moderate increase in nuclear shape irregularity, without indicating major tissue lesions (supplemental Figure 5A-B). We observed no alterations in the phosphorylation of AMPKα, Akt, and eNOS between the Btz and control groups (supplemental Figure 6E-F).
The Bip/GRP78 pathway is not involved in Cfz cardiotoxicity
Binding immunoglobulin protein (Bip; also referred to a glucose-regulated protein 78, GRP78) is a chaperone that is implicated in folding and assembly of proteins in the endoplasmic reticulum.21 PI induce GRP78 expression in myeloma cells.22 Because it has been demonstrated that Met suppresses GRP78, we evaluated the effects of Cfz and Btz and the coadministration of Met with Cfz on Bip/GRP78 pathway. We found that only Btz administration induced Bip/GRP78 expression (vs control), whereas neither Cfz nor Cfz+Met treatment altered the Bip/GRP78 expression (supplemental Figure 7).
Cfz increased PP2A phosphatase activity
To investigate the mechanism by which Cfz resulted in reduced phosphorylation of Akt and AMPKα in the myocardium without altering PTEN expression, we investigated the effects of the treatments on PP2A phosphatase activity. In protocols 2 and 3, Cfz induced an increase in PP2A activity compared with control, whereas in protocol 5A the activity of PP2A tended to show a slight (nonsignificant) increase; these findings were in parallel with the reduction of FS% observed in protocols 2 and 3 by Cfz. Met coadministration reduced PP2A activity in protocol 6A; however, no significant alteration was observed in protocol 6B (Figure 6A). We found no alterations in PP2A expression levels in any of the protocols used (Figure 6B-F).
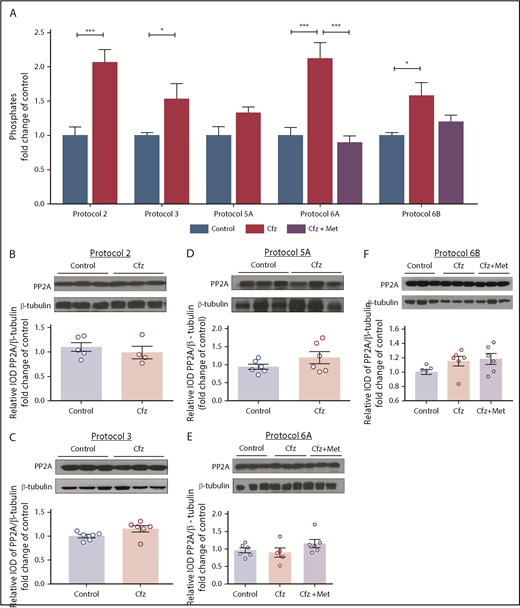
PP2A as a main mediator of Cfz-induced cardiotoxicity. (A) Graph of PP2A activity (pmol/min per milligram protein/mL) is expressed as fold change in comparison with controls. Representative western blot and relative densitometric analysis of PP2A in (B) protocol 2, (C) protocol 3, (D) protocol 5A, (E) protocol 6A, and (F) protocol 6B. *P < .05, **P < .01, ***P < .001 n = 4-6 per group. Western blots show 3 representative animals from each experimental group, 1 in each lane.
PP2A as a main mediator of Cfz-induced cardiotoxicity. (A) Graph of PP2A activity (pmol/min per milligram protein/mL) is expressed as fold change in comparison with controls. Representative western blot and relative densitometric analysis of PP2A in (B) protocol 2, (C) protocol 3, (D) protocol 5A, (E) protocol 6A, and (F) protocol 6B. *P < .05, **P < .01, ***P < .001 n = 4-6 per group. Western blots show 3 representative animals from each experimental group, 1 in each lane.
Discussion
Several PI exert some type of cardiotoxicity, with different severity indicating a possible class effect.23 Between Btz and Cfz, the latter demonstrated higher rates of cardiovascular adverse effects in comparison with Btz or other treatments in the setting of relapsed MM.23,24 Therefore, molecular-based evidence on using novel pharmacological interventions as prophylactic therapies against Cfz cardiotoxicity is of particular importance. The present in vivo study is the first to establish a translational protocol that correlates with the clinical observations in patients undergoing Cfz treatment. Cfz adversely affected myocardium through inactivation of AMPKα and downregulation of autophagy-related proteins when it was administered for 2 consecutive days. Alternative administration of Cfz for 6 days additionally resulted in inactivation of the PI3K/Akt/eNOS pathway. Our results highlight the pivotal role of PP2A activity in Cfz-induced cardiotoxicity and its downstream target, AMPKα. The critical role of AMPKα in Cfz-mediated cardiotoxicity was also evident by our finding that coadministration of metformin largely reversed the Cfz-induced molecular changes and restored cardiac function
To the best of our knowledge, there is no study conducted in animals to investigate Cfz effects on cardiac function with echocardiographic assessment. In the current guidelines, Cfz initial dosing is selected to be 27 or 56 mg/m2.1,5 The dose regimen might be altered upon manifestation of life-threatening cardiotoxicity and be reduced to 15 mg/m2 before medication discontinuation.25 Therefore, our experimental protocols provide a wide gamma of different dosing side by side with their functional and molecular effects. In a translational scope, the dose regimens selected for protocols 2, 3, and 4 are directly equivalent to the clinical praxis. In protocol 2, the total dose is equivalent to a human equivalent dose (HED) of 14.85 mg/m,2,26 which is the lowest dose used in MM patients. The dose of 8 mg/kg on alternate days (protocol 3) is equivalent to a HED of 29.65 mg/m2, whereas the dose of protocol 4 is equivalent to a HED of 59.3 mg/m2.26 Therefore, all dose regimens used in this study are in line with the dose regimens used in MM treatment in humans. However, metabolism and drug clearance in mice differs in comparison with humans27 ; therefore, more frequent administration of Cfz is needed to maintain the same effects as in humans, which is proven by the sustained proteasome inhibition in both myocardial tissue and PBMCs. Specifically, the inhibitory effect in PBMCs was relevant to inhibition observed in time courses of proteasome activity in patients.28,29 We conclusively established experimental protocols of translational value.
Interestingly, we observed that the FS% tends to decrease stepwise from a single dose to 2 doses of Cfz treatment. Four injections resulted in a moderate contractile dysfunction and LV dilation. Such changes are consistent with cardiac impairment and reduced ejection fraction in patients undergoing Cfz treatment30 and clinical cases reporting LV dilation upon Cfz administration for relapsed/refractory MM disease.31 Histology did not prove major myocardial lesions in Cfz-treated mice. Existing histological evidence on Cfz in rats are conflicting, indicating either myocardial degeneration combined with an increase in markers of myocardial tissue damage13,15 or negligible changes, with no alterations in pulmonary arteries wall thickness even in higher doses.32 Our results are in accordance with patients’ results indicating reversible cardiac dysfunction without necrosis or elevation of serum troponin levels.9 Cfz-induced cardiotoxicity is acute and can be reversible a few days after ending drug administration. When we stopped Cfz treatment, there was reversibility of the cardiac function and damage as evaluated by echocardiography because of the wash-out period of Cfz in intermittent, acute, and subchronic protocols.
By evaluating the molecular mechanisms of Cfz-induced cardiotoxicity, we focused first on molecular pathways that play pivotal roles in myocardial cell growth and survival, such as the PI3K/Akt/eNOS axis,33 and on signaling molecules related to autophagy, because cardiac-specific loss of autophagy causes cardiomyopathy in mice.34 Additionally, an inverse relationship between the ubiquitin–proteasome pathway and autophagy has been confirmed in different manifestations of cardiovascular disease.35 Based on our findings, Cfz inactivated PI3K/Akt/eNOS signaling only when administered for 4 alternative days; this effect was not PTEN-mediated as was shown for Btz.36 Cfz reduced eNOS phosphorylation; this effect correlated with iNOS induction. These molecular readouts indicate a deregulation of NO production37 ; a similar imbalance of eNOS/iNOS function has been described for doxorubicin-induced cardiotoxicity.38,39 Our findings are in agreement with cases in which PI produced impaired eNOS/NO function.40
Subsequently, we focused on signaling molecules related to the autophagy. In vivo studies have demonstrated that autophagy ablation leads to contractile dysfunction and LV dilation,34 findings with a similar pattern to the echocardiographic results of our study. AMPKα, besides regulating cardiac metabolism and energetics, modulates autophagic degradation through inhibition of mTOR complex 1.41,42 Cfz-induced inactivation of AMPKα in both protocols 2 and 3; this led to reduced Raptor phosphorylation, which has an inhibitory effect on mTOR signaling, and decreased the expression of the autophagic marker, LC3-II. Taken together, our results highlight that inactivation of AMPKα and downregulation of autophagy-related proteins occurred rapidly after Cfz administration. This seems to be the main mechanism of Cfz-induced cardiotoxicity (Figure 7); therefore, our next step focused on the upstream signaling of AMPKα.
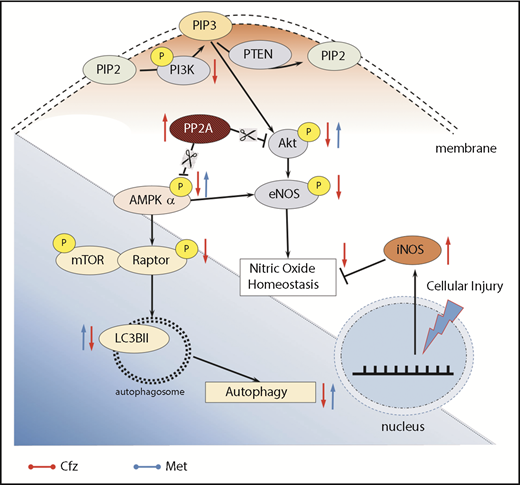
Proposed molecular mechanisms. Red arrows, Cfz-induced cardiotoxicity changes; blue arrows, Met-induced cardioprotection changes. P, phosphorylation.
Proposed molecular mechanisms. Red arrows, Cfz-induced cardiotoxicity changes; blue arrows, Met-induced cardioprotection changes. P, phosphorylation.
Reportedly, Cfz induced cell apoptosis by PP2A activation and pAkt dephosphorylation, independently of proteasome inhibition, through disturbing the expression of the oncoprotein KIAA1524, which inhibits PP2A.43 Our findings support the notion that Cfz activates PP2A (upstream suppressor of both Akt and AMPKα) and, plausibly, this effect justifies AMPKα inactivation. We observed no significant differences between the PP2A activity in protocol 5A, indicating that reversibility of the damage by Cfz depends on an off-target effect of the drug, which is the increased activity of PP2A.
Our next step was to confirm if the reduced phosphorylation of AMPKα is an essential molecular target of Cfz-induced cardiotoxicity. Met is an AMPKα activator18 and exerts cardioprotective effects mediated by autophagy regulation and apoptosis inhibition.44,45 The appropriate dose regimen in our study was selected based on the cardioprotective effects of Met in clinical trials.46,47 Therefore, we calculated the mouse-equivalent dose based on 850 mg Met hydrochloride twice daily with the use of interspecies dose extrapolation equations.48 Met coadministration with Cfz only for 2 days restored the cardiac function; this was accompanied by increased phosphorylation of AMPKα. Coadministration of Met for a longer period reversed Cfz-induced contractile dysfunction without exerting any glucose-lowering actions. Importantly, it did not interfere with the Cfz inhibitory effect on proteasome activity. At a molecular basis, the cardioprotective mechanism is facilitated through restoration of the AMPKα phosphorylation. Met restored mTOR complex 1 inhibition, enhancing constitutive autophagy in the heart, as demonstrated by the autophagic marker, LC3-II. The amplification of the autophagic flux is important for both attenuating proteasome inhibition-induced toxicity and preserving cellular viability in failing hearts.49,50 Especially in cardiomyocytes, autophagy is essential for maintaining cellular contractility because protein aggregates can intervene in the mechanical characteristics of the entire cell.51,52 Therefore, upon proteasome insufficiency to degrade misfolded proteins or aggregates, autophagy promotes cell survival. Met administration also resulted in activation of Akt. However, although Met protects diabetic endothelium through enhancing eNOS signaling,53,54 in our study, the coadministration of Met did not result in activation of eNOS and decreased expression of iNOS. Thus, the protection of the endothelium induced-cardiotoxicity by Cfz can be evaluated in further studies.
Met has been reported as the only antidiabetic drug to reduce cancer risk.55 Indeed, besides preclinical data supporting this hypothesis, there are multiple clinical trials investigating its anticancer potency.56 In the MM setting, diabetic patients receiving Met had increased overall survival compared with nonusers.57 Adding to this, Met inhibits disease progression of monoclonal gammopathy of undetermined significance and transformation to MM in patients with diabetes.58 Mechanism of action focuses on AMPKα-mediated induction of autophagy and cell-cycle arrest, leading to inhibition of proliferation in human MM cell lines and a murine xenograft myeloma model.59 The hypothesis that metformin enhances the apoptotic activity of Cfz will be the aim of our future studies. Despite the fact that synergistic activity of Met with Btz is not established, there is evidence supporting that Met could overcome Btz resistance through alteration of the unfolded protein response and autophagy.22,60 More specifically, Met cotreatment with Btz suppressed induction of the critical unfolded protein response effector GRP78 protein to impair autophagosome formation and enhance apoptosis.22 Our data comply with the literature, because only Btz could increase Bip expression in the myocardial tissue, an effect not evident in the Cfz group; Bip activation seems to be a drug effect of Btz not shared with Cfz.
A limitation of our study is that we evaluated healthy mice and not mice with a heart disease, which is the case in several elderly myeloma patients. Additionally, it has been reported that there is an age-dependent decline of proteasome functionality in rat hearts that was associated with decreased 20S proteasome content and loss of specific activities.61 Proteasomal dysfunction in the aging heart plays a role in myocardial ischemia and congestive heart failure.62 Given these findings, and considering that loss in proteasome function may impair the ability of myocytes to mount an appropriate response to stress, we speculate that irreversible proteasome inhibitors will result in enhanced susceptibility of the aged heart to cardiovascular disease. The aim of the current study was to clarify the underlying molecular mechanism of Cfz-induced cardiotoxicity independently of the existing comorbidities and aging, which will be the aim of our future scientific work. By investigating the molecular changes and knowing the molecular cause of this cardiotoxicity, we may use prophylactic therapy in patients, especially in those with comorbidities in a clinical study; therefore, it is important that we disclose here the prophylactic role of metformin. It is also important that subchronic administration of Cfz resulted in LV dilatation, but, surprisingly, the overall cardiac function was not significantly affected. The mechanism of this can also be the subject of further work.
In conclusion, our results provide the first evidence that the myocardial activation of PP2A by Cfz and the subsequent inactivation of AMPKα and the downstream signaling related to autophagy is an essential mechanism of the acute cardiac dysfunction of Cfz. Additionally, our study highlights Met as a cardioprotective agent during Cfz administration, and a clinical trial with the use of Met in patients who are treated with Cfz can be supported by these data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by a grant from the EU-CARDIOPROTECTION COST-ACTION (CA16225), a fellowship from the State Scholarships Foundation, Greece (E.-D.P.), and funding from the TASCMAR (EU-H2020/634674) and BIOIMAGING-GR (MIS 5002755) projects (I.P.T.).
Authorship
Contribution: P.E., G.K., I.A., and E.T. conceived and designed the study, performed the experiments, analyzed the data, and wrote the manuscript; P.E., G.K., A.V., P.-E.N., C.H.D., M.T., G.A., A.K., E.-D.P., I.P.T., and Z.K. performed the experiments, analyzed the data, and participated in the manuscript preparation; E.K., E.K.I., A.K., and M.A.D. participated in manuscript preparation and in the critical revision of the manuscript; and I.A. and E.T. supervised the study, analyzed the data, and revised the manuscript.
Conflict-of-interest disclosure: E.K., M.A.D., and E.T. have received honoraria from Amgen and Janssen outside the scope of this work. The remaining authors declare no competing financial interests.
Correspondence: Ioanna Andreadou, Faculty of Pharmacy, Panepistimiopolis, Zografou, Athens 15771, Greece; e-mail: jandread@pharm.uoa.gr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal