

In this issue of Blood, report using an animal model to study carfilzomib (Cfz)–induced cardiotoxicity and to demonstrate that the molecular mechanism is not related to the inhibition of the proteasome function but instead has an influence on modulation of the autophagy pathway, inactivation of AMPKα, and upregulation of PP2A phosphatase activity.1
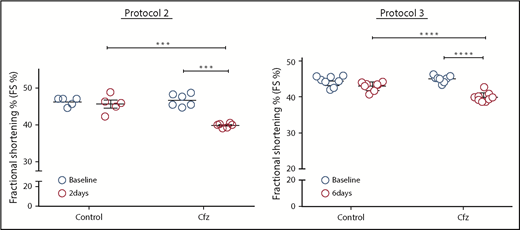
This figure demonstrates evidence of significant fractional shortening in all mice receiving Cfz without much variability. In protocol 2, the Cfz dose was equivalent to a human dose of 14.85 mg/m2 and was given for 2 days. In protocol 3, the Cfz dose was equivalent to a human dose of 29.65 mg/m2 given every other day for 4 doses. ***P = .001, ****P < .001. The figure was adapted from Figure 1E-F in the article by Efentakis et al that begins on page 710.
This figure demonstrates evidence of significant fractional shortening in all mice receiving Cfz without much variability. In protocol 2, the Cfz dose was equivalent to a human dose of 14.85 mg/m2 and was given for 2 days. In protocol 3, the Cfz dose was equivalent to a human dose of 29.65 mg/m2 given every other day for 4 doses. ***P = .001, ****P < .001. The figure was adapted from Figure 1E-F in the article by Efentakis et al that begins on page 710.
For many years, cardiotoxicity in patients with cancer has been associated with cumulative doses of anthracyclines. Then different types of cardiotoxicity caused by HER2/neu inhibitors emerged, and in recent years, with the advancement of targeted therapy in cancer, more and more agents have been reported to have cardiovascular adverse effects2 that necessitated the establishment of a new discipline of cardio-oncology.
Carfilzomib is an irreversible proteasome inhibitor that has been shown to be effective in the treatment of relapsed multiple myeloma. However, it has been shown to have significant grade 3 to 4 cardiotoxicity in as many as 5% to 10% of patients.3 This incidence is higher than that reported for bortezomib, although cardiotoxicity has been reported with all clinically approved proteasome inhibitors.4 Fortunately, Cfz-induced cardiotoxicity is reversible in many patients after the drug is stopped. Several studies have tried to assess clinical predictive factors, but the occurrence of symptomatic Cfz-induced cardiotoxicity is still mostly unpredictable. Thus, it is important to study its pathogenesis, predictive biomarkers, and ways to protect patients against such toxicity.
The article by Efentakis et al focuses on establishing a clinically relevant mouse model to study the cardiotoxic effects of Cfz by echocardiography and correlating the findings with changes in molecular targets. By using C57BL/6 male mice, the authors established appropriate Cfz treatment regimens that resulted in cardiotoxicity that can be reversible upon stopping the drug but can also be prevented by cotreatment with metformin (Met). The surprising finding is the off-target effects of Cfz on autophagy proteins and activation of PPA2 (the reasons for the cardiotoxicity) and not proteasome inhibition. Thus, Met (a known activator of MAPKα and promoter of autophagy, as reported by many studies) has protective effects without interfering with proteasome inhibition. Conversely, the administration of bortezomib in this animal model did not result in cardiotoxicity or in the molecular changes observed with Cfz.
Cardiotoxicity animal models have been used to elucidate possible molecular mechanisms and demonstrate protective effects of different agents, including Met. A quick search of PubMed using the 3-word phrase “cardiotoxicity animal models” revealed 584 publications on the topic, and most dealt with anthracycline-induced cardiotoxicity. One study published in 20175 used a Wistar albino male rat model to study Cfz cardiotoxicity, which was measured by histopathologic changes as well as molecular and biochemical markers. In that study, rutin (a bioflavonoid) was able to reverse Cfz-induced cardiotoxicity. The molecular and biochemical studies pointed to the NF-κB pathway, the cardiac hypertrophic gene, and oxidative stress as being responsible for Cfz-induced toxicity. These findings raise a question about whether using different animal models is applicable for studying effects in humans. The real test will come when clinical trials confirm the findings of these animal studies; results and conclusions remain to be seen.
Another issue regarding whether studies in animal models are applicable to humans is related to the fact that not all patients receiving Cfz will develop cardiotoxicity, but all animals in these models will develop cardiotoxicity (see figure). Other factors such as the effects of old age and prior heart disease in patients could explain the difference. Genetic polymorphism could also be a factor that plays a role in different patients’ susceptibility, and it needs to be investigated. There is a body of literature on using pharmacogenomics in anthracycline-induced cardiotoxicity6,7 that led the Canadian Pharmacogenomics Network for Drug Safety to recommend genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity.8 Genetic risk profiling could be used to identify high-risk patients who can then be provided with safer treatment options, including cardioprotective agents.
From the literature on Met, one gets the impression that it is a wonder drug because of its potential beneficial effects in cancer treatment and its lack of significant adverse effects. The successful use of Met to protect against Cfz-induced cardiotoxicity could be a breakthrough in the field, and it will have major impact on treatment for patients with multiple myeloma. Autophagy is a complex process involved in cell homeostasis, and novel modulators are being actively pursued.9 Once novel modulators with a better therapeutic index are discovered, their cardioprotective effects can be tested by using the animal model presented by Efentakis et al.
Overall, the study by Efentakis et al provides new insights into the mechanism of Cfz-induced cardiotoxicity by establishing a relevant animal model and demonstrates the protective effects of Met. But a few questions remain to be answered. In addition to those raised by the authors in the “Discussion,” there are other questions of interest. Because all proteasome inhibitors are reported to cause some cardiotoxicity but less cardiotoxicity than Cfz, what is the mechanism for the other proteasome inhibitor–induced cardiotoxicities and how can the higher incidence of cardiotoxicity with Cfz be explained? Are the animal data from this study or any other animal model going to be clinically applicable to human patients? Clinical trials in humans are needed to answer this question. And last, will the recently approved dose and dosing schedule for Cfz be less or more cardiotoxic?
Conflict-of-interest disclosure: J.S.M. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal