

Key Points
GM-CSF neutralization with lenzilumab during CART19 therapy prevents CRS and neuroinflammation and is a viable therapeutic solution.
GM-CSF neutralization and GM-CSF knockout in CAR-T cells enhance their antitumor functions.

Abstract
Chimeric antigen receptor T (CAR-T) cell therapy is a new pillar in cancer therapeutics; however, its application is limited by the associated toxicities. These include cytokine release syndrome (CRS) and neurotoxicity. Although the IL-6R antagonist tocilizumab is approved for treatment of CRS, there is no approved treatment of neurotoxicity associated with CD19-targeted CAR-T (CART19) cell therapy. Recent data suggest that monocytes and macrophages contribute to the development of CRS and neurotoxicity after CAR-T cell therapy. Therefore, we investigated neutralizing granulocyte-macrophage colony-stimulating factor (GM-CSF) as a potential strategy to manage CART19 cell–associated toxicities. In this study, we show that GM-CSF neutralization with lenzilumab does not inhibit CART19 cell function in vitro or in vivo. Moreover, CART19 cell proliferation was enhanced and durable control of leukemic disease was maintained better in patient-derived xenografts after GM-CSF neutralization with lenzilumab. In a patient acute lymphoblastic leukemia xenograft model of CRS and neuroinflammation (NI), GM-CSF neutralization resulted in a reduction of myeloid and T cell infiltration in the central nervous system and a significant reduction in NI and prevention of CRS. Finally, we generated GM-CSF–deficient CART19 cells through CRISPR/Cas9 disruption of GM-CSF during CAR-T cell manufacturing. These GM-CSFk/o CAR-T cells maintained normal functions and had enhanced antitumor activity in vivo, as well as improved overall survival, compared with CART19 cells. Together, these studies illuminate a novel approach to abrogate NI and CRS through GM-CSF neutralization, which may potentially enhance CAR-T cell function. Phase 2 studies with lenzilumab in combination with CART19 cell therapy are planned.
Introduction
Chimeric antigen receptor T (CAR-T) cell therapy has emerged as a novel and potentially revolutionary therapy to treat cancer.1,2 Based on unprecedented responses in B cell malignancies, 2 CD19-targeted CAR-T (CART19) cell products were approved by the US Food and Drug Administration in 2017.3-5 However, the wider application of CAR-T cell therapy is limited by the emergence of unique and potentially fatal toxicities. These include the development of cytokine release syndrome (CRS) and neurotoxicity.3,5-7 Up to 50% of patients treated with CART19 cells develop grade 3 or higher CRS or neurotoxicity, and several deaths have been reported.3,4,8 These toxicities are associated with prolonged hospitalization and intensive care unit stays,9 and the long-term effects of neurotoxicity are unknown. Thus, controlling these CART19 cell–related toxicities is imperative to lessen morbidity, mortality, duration of hospitalization, intensive care unit admissions, the supportive care required, and the significant indirect costs associated with CAR-T cell therapy.
The development of CRS is directly related to in vivo T-cell expansion and massive production of T-cell effector cytokines (eg, interleukin-6 [IL-6], interferon-γ [IFN-γ], monocyte chemoattractant protein 1 [MCP-1], and granulocyte-macrophage colony-stimulating factor [GM-CSF]).3,4,10,11 Although neurotoxicity is also associated with elevation of several key cytokines that follow CRS development,12,13 the exact mechanism for neurotoxicity development is unknown. Recent data indicate that CD14+ monocytes are significantly increased in the cerebrospinal fluid (CSF) from patients who developed grade 3 or 4 neurotoxicity after CART19 cell therapy.14 Serum levels of GM-CSF, ferritin, and IL-2 were the markers associated with the development of neurotoxicity, with GM-CSF being the most significantly associated with the development of grade 3 or 4 neurotoxicity based on correlative studies from the ZUMA-1 pivotal trial of CART19 cell treatment in diffuse large B-cell lymphoma.3 The early elevation of myeloid-differentiating chemokines (eg, MIP-1α and MCP-1) was predictive for the development of severe CRS in algorithms developed after treatment of pediatric patients with CART19 cell therapy.10 In addition, preclinical experiments have shown that IL-6, a key cytokine in the development of CRS, is not produced by CAR-T cells; rather, it is predominantly produced by monocytes and macrophages.15 Collectively, these results suggest a potential role for GM-CSF and myeloid cells in the development of CRS and neurotoxicity.
There are no effective therapies for the prevention of CRS or neurotoxicity, and the only available treatment for severe neurotoxicity is high-dose corticosteroids. However, there is no supportive evidence that corticosteroids improve neurotoxicity, and the early administration of corticosteroids may interfere with CAR-T effector functions. Tocilizumab, an IL-6 receptor antagonist, is effective and is approved by the US Food and Drug Administration for the treatment of severe CRS.3-5,7 Although tocilizumab was a critical development in the birth of the field, severe toxicities and death still occur from CRS, despite its routine use.16 Importantly, tocilizumab is not an approved treatment for neurotoxicity associated with CART19 cell therapy.17,18 In an analysis from the ZUMA-1 clinical trial, the overall rates of neurotoxicity and the rates of grade ≥ 3 neurotoxicity were increased with early prophylactic tocilizumab, which is believed to be driven by the transient increase in IL-6 levels seen following the initiation of tocilizumab.3,14,19
These observations provide a solid rationale to investigate neutralization of GM-CSF as a potential strategy to abrogate CRS and neuroinflammation (NI) associated with CAR-T cell therapy. Lenzilumab is a monoclonal antibody that neutralizes human GM-CSF. More than 100 patients have been treated with this antibody in multiple single- and repeat-dose phase 1 and 2 clinical trials, including a phase 1 study in healthy volunteers, a safety run-in to a phase 2 study in subjects with moderate to severe rheumatoid arthritis (terminated early because of development plan refocus), and a phase 2 study in subjects with asthma. Across these clinical trials, lenzilumab was found to be safe and well tolerated, with no grade >2 adverse events reported and no evidence of granulocytopenia (absolute neutrophil count <1000 per microliter), monocytopenia, or severe infusion-related reactions. All events were self-limiting or were treated with medication and resolved without sequelae.20-22 An additional phase 1 clinical trial is ongoing in subjects with chronic myelomonocytic leukemia (NCT02546284).
Here, we propose a new paradigm in the management of CRS and NI associated with CAR-T cell therapy via the inhibition of GM-CSF as a preventative strategy. We found that the GM-CSF–neutralizing antibody lenzilumab does not inhibit CAR-T cell function, and it may enhance their antitumor activity. GM-CSF neutralization prevented CRS and significantly reduced the severity of NI in a patient acute lymphoblastic leukemia (ALL) patient-derived xenograft model of CRS and NI post-CART19 cell therapy. Finally, we show that GM-CSF disruption during CAR-T cell manufacturing results in GM-CSFk/o CAR-T cells that continue to exhibit potent effector functions, despite reduced production of GM-CSF. This provides evidence for the clinical feasibility of combining CAR-T cell therapy with the GM-CSF–neutralizing antibody lenzilumab and the potential to develop CAR-T cells that express reduced GM-CSF levels.
Materials and methods
The materials and methods used in this study are described in Supplemental methods (available on the Blood Web site).
Results
GM-CSF neutralization in vitro enhances CAR-T cell proliferation in the presence of monocytes and does not impair CAR-T cell effector function
If GM-CSF neutralization after CAR-T cell therapy is to be used as a strategy to prevent CRS and neurotoxicity, it must not inhibit CAR-T cell efficacy. Therefore, our initial experiments aimed to investigate the impact of GM-CSF neutralization on CAR-T cell effector functions. CART19 cells were cocultured or not with the CD19+ ALL cell line NALM6 in the presence of lenzilumab (GM-CSF–neutralizing antibody) or an isotype control (immunoglobulin G [IgG]). We established that lenzilumab, but not IgG control antibody, was indeed able to completely neutralize GM-CSF (Figure 1A) but did not inhibit CAR-T cell antigen-specific proliferation (Figure 1B). When CART19 cells were cocultured with the CD19+ cell line NALM6 in the presence of monocytes, lenzilumab in combination with CART19 cells demonstrated an exponential increase in antigen-specific CART19 cell proliferation compared with CART19 cells plus isotype control IgG (P < .0001, Figure 1C). To investigate CAR-T–specific cytotoxicity, CART19 cells or control untransduced (UTD) T cells were cultured with the luciferase+CD19+ NALM6 cell line and treated with isotype-control antibody or GM-CSF–neutralizing antibody (Figure 1D). GM-CSF–neutralizing antibody treatment did not inhibit the ability of CAR-T cells to kill NALM6 target cells (Figure 1D). Overall, these results indicate that lenzilumab does not inhibit CAR-T cell function in vitro and enhances CART19 cell proliferation in the presence of monocytes, suggesting that GM-CSF neutralization may improve CAR-T cell–mediated efficacy.

GM-CSF neutralization in vitro enhances CAR-T cell proliferation in the presence of monocytes and does not impair CAR-T–cell effector function. (A) Lenzilumab neutralizes CAR-T cell–produced GM-CSF in vitro compared with isotype-control treatment, as assayed by a multiplex assay after 3 days of culture with CART19 cells in media alone or CART19 cells cocultured with NALM6; n = 2 experiments, 2 replicates per experiment, a representative experiment is depicted. (B) GM-CSF–neutralizing antibody treatment did not inhibit the ability of CAR-T cells to proliferate, as assayed by a carboxyfluorescein diacetate succinimidyl ester flow cytometry proliferation assay of live CD3 cells; n = 3 donors, 2 replicates per donor, a representative experiment at the 3-day time point is depicted. (C) Lenzilumab enhanced the proliferation of CART19 cells compared with isotype-control–treated with CART19 cells when cocultured with monocytes; n = 3 donors at the 3-day time point, 2 replicates per donor. (D) Lenzilumab treatment did not inhibit cytotoxicity of CART19 cells or UTD T cells when cultured with NALM6; n = 3 donors, 2 replicates per donor, a representative experiment at the 48-hour time point is depicted. All data are mean ± SEM. ***P < .001, ****P < .0001, Student t test. Alone, CART19 cells in media alone; MOLM13, CART19+MOLM13; ns, P > .05, lenzilumab vs isotype-control treatment using the Student t test; NALM6, CART19+NALM6; PMA/ION, CART19 cells plus 5 ng/mL PMA and 0.1 μg/mL ionomycin.
GM-CSF neutralization in vitro enhances CAR-T cell proliferation in the presence of monocytes and does not impair CAR-T–cell effector function. (A) Lenzilumab neutralizes CAR-T cell–produced GM-CSF in vitro compared with isotype-control treatment, as assayed by a multiplex assay after 3 days of culture with CART19 cells in media alone or CART19 cells cocultured with NALM6; n = 2 experiments, 2 replicates per experiment, a representative experiment is depicted. (B) GM-CSF–neutralizing antibody treatment did not inhibit the ability of CAR-T cells to proliferate, as assayed by a carboxyfluorescein diacetate succinimidyl ester flow cytometry proliferation assay of live CD3 cells; n = 3 donors, 2 replicates per donor, a representative experiment at the 3-day time point is depicted. (C) Lenzilumab enhanced the proliferation of CART19 cells compared with isotype-control–treated with CART19 cells when cocultured with monocytes; n = 3 donors at the 3-day time point, 2 replicates per donor. (D) Lenzilumab treatment did not inhibit cytotoxicity of CART19 cells or UTD T cells when cultured with NALM6; n = 3 donors, 2 replicates per donor, a representative experiment at the 48-hour time point is depicted. All data are mean ± SEM. ***P < .001, ****P < .0001, Student t test. Alone, CART19 cells in media alone; MOLM13, CART19+MOLM13; ns, P > .05, lenzilumab vs isotype-control treatment using the Student t test; NALM6, CART19+NALM6; PMA/ION, CART19 cells plus 5 ng/mL PMA and 0.1 μg/mL ionomycin.
GM-CSF neutralization in vivo enhances CAR-T cell antitumor activity in xenograft models
To confirm that GM-CSF depletion does not inhibit CART19 cell effector functions, we investigated the role of GM-CSF neutralization with lenzilumab on CART19 cell antitumor activity in xenograft models. First, we used a relapse model intended to vigorously investigate whether the antitumor activity of CART19 cells was impacted by GM-CSF neutralization. NOD/SCID/IL-2 receptor γ null (NSG) mice were injected with 1 × 106 luciferase+ NALM6 cells and imaged 6 days later, allowing sufficient time for mice to achieve very high tumor burdens. Mice were randomized to receive a single injection of CART19 or UTD T cells and 10 days of isotype-control antibody or lenzilumab (Figure 2A). GM-CSF assay on serum collected 8 days after CART19 cell injection revealed that lenzilumab successfully neutralizes GM-CSF in the context of CART19 cell therapy (Figure 2B). Bioluminescence imaging 1 week after CART19 cell injection showed that CART19 cells, in combination with lenzilumab, effectively controlled leukemia in this high tumor burden relapse model and did so significantly better than control UTD T cells (Figure 2C-D). Treatment with CART19 cells in combination with lenzilumab resulted in potent antitumor activity and improved overall survival, similar to CART19 cells with control antibody despite neutralization of GM-CSF levels, indicating that GM-CSF does not impair CAR-T cell activity in vivo (supplemental Figure 3). Second, we performed these experiments in an ALL patient–derived xenograft model, in the presence of human peripheral blood mononuclear cells, because this represents a more relevant heterogeneous model. After conditioning chemotherapy with busulfan, mice were injected with blasts derived from patients with relapsed ALL. Mice were monitored for engraftment for several weeks through serial tail vein bleedings. When the CD19+ blasts in the blood were ∼1 per microliter, mice were randomized to receive CART19 cell treatment, in combination with peripheral blood mononuclear cells with either lenzilumab plus an anti-mouse GM-CSF–neutralization antibody or isotype-control IgG antibodies starting on the day of CART19 cell injection for 10 days (Figure 2E). In this patient ALL xenograft model, GM-CSF neutralization, in combination with CART19 cell therapy, resulted in a significant improvement in leukemic disease control sustained over time for ≥35 days post–CART19 cell administration compared with CART19 cells plus isotype control (Figure 2F). This suggests that GM-CSF neutralization may play a role in reducing relapses and increasing durable complete responses after CART19 cell therapy.
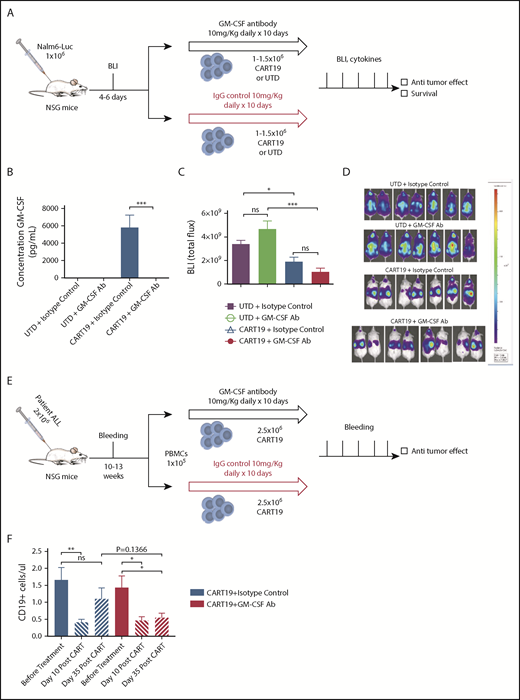
GM-CSF neutralization in vivo enhances CAR-T cell antitumor activity in xenograft models. (A) Experimental schema: NSG mice were injected IV with the luciferase+CD19+ cell line NALM6 (1 × 106 cells per mouse). Four to six days later, mice were imaged and randomized, and they received 1 to 1.5 × 106 CART19 cells or an equivalent number of total cells of control UTD cells the following day, with lenzilumab or control IgG (10 mg/kg, given intraperitoneally daily for 10 days, starting on the day of CAR-T injection). Mice were followed with serial bioluminescence imaging to assess disease burden beginning at day 7 post–CAR-T cell injection and were followed for overall survival. Tail vein bleeding was performed 7 or 8 days after CAR-T cell injection. (B) Lenzilumab neutralizes CAR-T–produced serum GM-CSF in vivo compared with isotype-control treatment, as assayed by a GM-CSF singleplex assay; n = 2 experiments, 7 or 8 mice per group, representative experiment, serum from day 8 post–CAR-T cell/UTD cell injection. Data are mean ± SEM. (C) Lenzilumab-treated CAR-T cells and isotype-control–treated CAR-T cells are equally effective at controlling tumor burden in vivo in a high tumor burden relapse xenograft model of ALL, day 7 post–CAR-T injection; n = 2 experiments, 7 or 8 mice per group, a representative experiment is depicted. Data are mean ± SEM. (D) Mouse images from (C). (E) Experimental schema: NSG mice were injected IV with the blasts derived from patients with ALL (1 × 106 cells per mouse). Mice were bled serially and when the CD19+ cells were ∼1 per microliter, mice were randomized to receive 2.5 × 106 CART19 cells with lenzilumab or control IgG (10 mg/kg, given intraperitoneally daily for 10 days, starting on the day of CAR-T injection). Mice were followed with serial tail vein bleeding to assess disease burden beginning at day 14 post–CAR-T cell injection and were followed for overall survival. (F) Lenzilumab treatment with CAR-T therapy results in more sustained control of tumor burden over time in a patient ALL xenograft model compared with isotype-control treatment with CAR-T therapy; 6 mice per group. Data are mean ± SEM. *P < .05, **P < .01, ***P < .001, Student t test. ns, P > .05, Student t test.
GM-CSF neutralization in vivo enhances CAR-T cell antitumor activity in xenograft models. (A) Experimental schema: NSG mice were injected IV with the luciferase+CD19+ cell line NALM6 (1 × 106 cells per mouse). Four to six days later, mice were imaged and randomized, and they received 1 to 1.5 × 106 CART19 cells or an equivalent number of total cells of control UTD cells the following day, with lenzilumab or control IgG (10 mg/kg, given intraperitoneally daily for 10 days, starting on the day of CAR-T injection). Mice were followed with serial bioluminescence imaging to assess disease burden beginning at day 7 post–CAR-T cell injection and were followed for overall survival. Tail vein bleeding was performed 7 or 8 days after CAR-T cell injection. (B) Lenzilumab neutralizes CAR-T–produced serum GM-CSF in vivo compared with isotype-control treatment, as assayed by a GM-CSF singleplex assay; n = 2 experiments, 7 or 8 mice per group, representative experiment, serum from day 8 post–CAR-T cell/UTD cell injection. Data are mean ± SEM. (C) Lenzilumab-treated CAR-T cells and isotype-control–treated CAR-T cells are equally effective at controlling tumor burden in vivo in a high tumor burden relapse xenograft model of ALL, day 7 post–CAR-T injection; n = 2 experiments, 7 or 8 mice per group, a representative experiment is depicted. Data are mean ± SEM. (D) Mouse images from (C). (E) Experimental schema: NSG mice were injected IV with the blasts derived from patients with ALL (1 × 106 cells per mouse). Mice were bled serially and when the CD19+ cells were ∼1 per microliter, mice were randomized to receive 2.5 × 106 CART19 cells with lenzilumab or control IgG (10 mg/kg, given intraperitoneally daily for 10 days, starting on the day of CAR-T injection). Mice were followed with serial tail vein bleeding to assess disease burden beginning at day 14 post–CAR-T cell injection and were followed for overall survival. (F) Lenzilumab treatment with CAR-T therapy results in more sustained control of tumor burden over time in a patient ALL xenograft model compared with isotype-control treatment with CAR-T therapy; 6 mice per group. Data are mean ± SEM. *P < .05, **P < .01, ***P < .001, Student t test. ns, P > .05, Student t test.
GM-CSF CRISPR-knockout CAR-T cells exhibit reduced expression of GM-CSF, similar levels of key cytokines and chemokines, and enhanced antitumor activity
To confidently exclude any critical role for GM-CSF in CAR-T cell function, we disrupted the GM-CSF gene during CAR-T cell manufacturing using a guide RNA that has been reported to yield high-efficiency knockout and is cloned into a CRISPR lentivirus backbone.23 Using this guide RNA, we achieved ∼60% knockout efficiency in CART19 cells (supplemental Figure 4). When CAR-T cells were stimulated with the CD19+ cell line NALM6, GM-CSFk/o CAR-T cells produced statistically significantly less GM-CSF compared with CART19 cells with a wild-type GM-CSF locus (wild-type CART19 cells). GM-CSF knockout in CAR-T cells did not impair the production of other key T-cell cytokines, including IFN-γ, IL-2, or CAR-T cell antigen–specific degranulation (CD107a) (Figure 3A), but it did exhibit reduced expression of GM-CSF (Figure 3B). To confirm that GM-CSFk/o CAR-T cells continue to exhibit normal functions, we tested their in vivo efficacy in the high tumor burden relapsing xenograft model of ALL (as described in Figure 2A). In this xenograft model, utilization of GM-CSFk/o CART19 cells instead of wild-type CART19 cells markedly reduced serum levels of human GM-CSF at 7 days after CART19 cell treatment (Figure 3B). Bioluminescence imaging data implied that GM-CSFk/o CART19 cells show enhanced leukemic control compared with CART19 cells in this model (supplemental Figure 5). Importantly, GM-CSFk/o CART19 cells demonstrated significant improvement in overall survival compared with wild-type CART19 cells (Figure 3C). Human GM-CSF was statistically significantly decreased (via Student t test) in GM-CSFk/o CART19 cells compared with wild-type CART19 cells (Figure 3D). Visually, the mouse GM-CSF appears to be increased, although this is not statistically significant via the Student t test (P = .472367) (Figure 3E). This lack of mouse GM-CSF reduction is not necessarily surprising, because GM-CSFk/o CART19 cells (which are human) are the only cells within the mouse that possess the knockout; thus, mouse GM-CSF likely would not be affected directly. By visual inspection, mouse IFN-γ–induced protein (IP-10), a chemokine that attracts numerous cell types, including T cells and monocytes, appears paradoxically increased in GM-CSFk/o CART19 cells compared with CART19 cells, but this is also not statistically significant (P = .4877, Student t test) (Figure 3E). By visual inspection, mouse MIP-1α (an inflammatory cytokine important in neutrophil attraction) and mouse macrophage colony-stimulating factor (a cytokine critical in macrophage differentiation) appear to be reduced, although they are not statistically significant (P = .2437 and P = .3619, respectively) (Figure 3E). Mouse IL-1b, a critical inflammatory cytokine produced by macrophages, and mouse IL-15, a cytokine produced by macrophages that aids in natural killer cell proliferation, appear to be reduced in GM-CSFk/o CART19 cells compared with CART19 cells (Figure 3E), with P = .0741 and P = .0900, respectively (Figure 3E). Critical human T-cell cytokines were not inhibited by GM-CSFk/o (Figure 3D). It should be emphasized that these xenografts were produced with high burdens of the NALM6 cell line, and our CRS/NI model (Figures 4-7) requires the use of patient ALL cells to be generated. Thus, unsurprisingly, cytokine profiles differ between the 2 models because the NALM6 xenografts (Figure 3) do not develop CRS or NI. Together, in the context of a NALM6 high tumor burden model without CRS, the results in Figure 3 confirm Figures 1 and 2, indicating that GM-CSF depletion does not impair normal cytokines or chemokines that are critical to CAR-T efficacy functions. In addition, the results in Figure 3 indicate that GM-CSFk/o CAR-T cells may represent a therapeutic option for “built-in” GM-CSF control as a modification during CAR-T cell manufacturing.

GM-CSF CRISPR-knockout CAR-T cells exhibit reduced expression of GM-CSF, similar levels of key cytokines and chemokines, and enhanced antitumor activity. (A) CRISPR Cas9 GM-CSFk/o CART19 cells exhibit reduced GM-CSF production compared with wild-type CART19 cells, but other cytokine production and degranulation are not inhibited by the GM-CSF gene disruption. CART19 cells and GM-CSFk/o CART19 cells stimulated with NALM6; n = 3 experiments, 2 replicates per experiment. Data are mean ± SEM. ***P < .001, Student t test. n.s., P > .05, Student t test. (B) GM-CSF k/o CAR-T cells have reduced serum human GM-CSF in vivo compared with CAR-T treatment, as assayed by multiplex; 5 or 6 mice per group (4 to 6 at time of bleed, 8 days post–CAR-T cell injection). Data are mean ± SEM. ***P < .001, ****P < .0001, Student t test. (C) GM-CSFk/o CART19 cells in vivo enhance overall survival compared with wild-type CART19 cells in a high tumor burden relapse xenograft model of ALL utilizing a NALM6 cell line; 5 or 6 mice per group. **P < .01, log-rank test. Human (D) and mouse (E) multiplex of serum cytokines and chemokines. A statistically significant reduction of human GM-CSF in GM-CSFk/o CART19 cells compared to wild-type CAR-T cells was observed, but no other statistically significant differences between GM-CSFk/o CART19 cells and wild-type CART19 cells were seen, further implicating that critical T-cell cytokines and chemokines are not adversely depleted by reducing GM-CSF expression; 5 or 6 mice per group (4 to 6 at time of bleed). ****P < .0001, Student t test.
GM-CSF CRISPR-knockout CAR-T cells exhibit reduced expression of GM-CSF, similar levels of key cytokines and chemokines, and enhanced antitumor activity. (A) CRISPR Cas9 GM-CSFk/o CART19 cells exhibit reduced GM-CSF production compared with wild-type CART19 cells, but other cytokine production and degranulation are not inhibited by the GM-CSF gene disruption. CART19 cells and GM-CSFk/o CART19 cells stimulated with NALM6; n = 3 experiments, 2 replicates per experiment. Data are mean ± SEM. ***P < .001, Student t test. n.s., P > .05, Student t test. (B) GM-CSF k/o CAR-T cells have reduced serum human GM-CSF in vivo compared with CAR-T treatment, as assayed by multiplex; 5 or 6 mice per group (4 to 6 at time of bleed, 8 days post–CAR-T cell injection). Data are mean ± SEM. ***P < .001, ****P < .0001, Student t test. (C) GM-CSFk/o CART19 cells in vivo enhance overall survival compared with wild-type CART19 cells in a high tumor burden relapse xenograft model of ALL utilizing a NALM6 cell line; 5 or 6 mice per group. **P < .01, log-rank test. Human (D) and mouse (E) multiplex of serum cytokines and chemokines. A statistically significant reduction of human GM-CSF in GM-CSFk/o CART19 cells compared to wild-type CAR-T cells was observed, but no other statistically significant differences between GM-CSFk/o CART19 cells and wild-type CART19 cells were seen, further implicating that critical T-cell cytokines and chemokines are not adversely depleted by reducing GM-CSF expression; 5 or 6 mice per group (4 to 6 at time of bleed). ****P < .0001, Student t test.
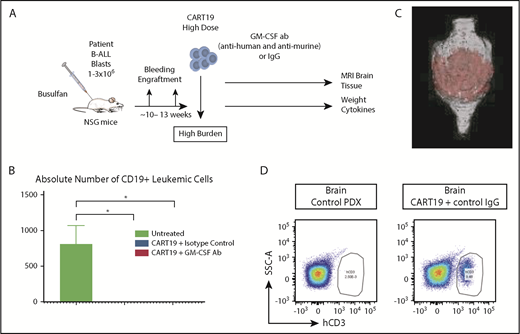
Patient-derived xenograft model for NI and CRS. (A) Experimental schema: mice received 1 to 3 × 106 blasts derived from the peripheral blood of patients with ALL. Mice were monitored for engraftment for ∼10 to 13 weeks via tail vein bleeding. When serum CD19+ cells were ≥10 per microliter, the mice received CART19 cells (2 to 5 × 106 cells) and commenced antibody therapy for a total of 10 days, as indicated. Mice were weighed on a daily basis as a measure of their well-being. Mouse brain MRIs were performed 5 or 6 days post–CART19 cell injection, and tail vein bleeding for cytokine/chemokine and T cell analysis was performed 4 to 11 days post–CART19 cell injection; 2 independent experiments. (B) Combination of GM-CSF neutralization with CART19 cells is equally effective as isotype control antibodies combined with CART19 cells in controlling CD19+ burden of ALL cells; representative experiment, 3 mice per group, 11 days post–CART19 cell injection. Data are mean ± SEM. (C) Brain MRI with CART19 cell therapy exhibits T1 enhancement, suggestive of blood–brain barrier disruption and possible edema; 3 mice per group, 5 or 6 days post–CART19 cell injection, representative image. (D) High tumor burden ALL patient–derived xenografts (PDX) treated with CART19 cells show human CD3 cell infiltration of the brain compared with untreated PDX controls; 3 mice per group, representative image. *P < .05, Student t test.
Patient-derived xenograft model for NI and CRS. (A) Experimental schema: mice received 1 to 3 × 106 blasts derived from the peripheral blood of patients with ALL. Mice were monitored for engraftment for ∼10 to 13 weeks via tail vein bleeding. When serum CD19+ cells were ≥10 per microliter, the mice received CART19 cells (2 to 5 × 106 cells) and commenced antibody therapy for a total of 10 days, as indicated. Mice were weighed on a daily basis as a measure of their well-being. Mouse brain MRIs were performed 5 or 6 days post–CART19 cell injection, and tail vein bleeding for cytokine/chemokine and T cell analysis was performed 4 to 11 days post–CART19 cell injection; 2 independent experiments. (B) Combination of GM-CSF neutralization with CART19 cells is equally effective as isotype control antibodies combined with CART19 cells in controlling CD19+ burden of ALL cells; representative experiment, 3 mice per group, 11 days post–CART19 cell injection. Data are mean ± SEM. (C) Brain MRI with CART19 cell therapy exhibits T1 enhancement, suggestive of blood–brain barrier disruption and possible edema; 3 mice per group, 5 or 6 days post–CART19 cell injection, representative image. (D) High tumor burden ALL patient–derived xenografts (PDX) treated with CART19 cells show human CD3 cell infiltration of the brain compared with untreated PDX controls; 3 mice per group, representative image. *P < .05, Student t test.

Canonical pathways are altered in brains from patient-derived xenografts after treatment with CART19 cells. Red boxes indicate upregulation of genes in CART19 cells plus isotype control–treated mice compared with the untreated patient-derived xenografts.
Canonical pathways are altered in brains from patient-derived xenografts after treatment with CART19 cells. Red boxes indicate upregulation of genes in CART19 cells plus isotype control–treated mice compared with the untreated patient-derived xenografts.

GM-CSF neutralization in vivo ameliorates CRS after CART19 therapy in a xenograft model. (A) Lenzilumab and anti-mouse GM-CSF antibody prevent CRS-induced weight loss compared with mice treated with CART19 cells and isotype-control antibodies; 3 mice per group, 2-way analysis of variance. Data are mean ± SEM. (B) Human GM-CSF was neutralized in patient-derived xenografts treated with lenzilumab and mouse GM-CSF–neutralizing antibody; 3 mice per group. Data are mean ± SEM. *P < .05, ***P < .001, Student t test. (C) Human cytokine/chemokine heat map (serum collected 11 days after CART19 cell injection) exhibits increases in cytokines and chemokines typical of CRS after CART19 cell treatment. GM-CSF neutralization results in a significant decrease in several cytokines and chemokines compared with mice treated with CART19 cells and isotype-control antibodies, including several myeloid-associated cytokines and chemokines, as indicated in the panel; 3 mice per group, serum from day 11 post–CART19 cell injection. *P < .05, **P < .01, ***P < .001, GM-CSF neutralizing antibody–treated mice vs isotype control–treated mice that received CAR-T cell therapy, Student t test. (D) Mouse cytokine/chemokine heat map (serum collected 11 days after CART19 cell injection) exhibits increases in mouse cytokines and chemokines typical of CRS after CART19 cell treatment. GM-CSF neutralization results in a significant decrease in several cytokines and chemokines compared with treatment with CART19 cells with control antibodies, including several myeloid-differentiating cytokines and chemokines, as indicated in the panel; 3 mice per group, serum from day 11 post–CART19 cell injection, *P < .05, GM-CSF neutralizing antibody–treated mice vs isotype control–treated mice that received CAR-T cell therapy, Student t test.
GM-CSF neutralization in vivo ameliorates CRS after CART19 therapy in a xenograft model. (A) Lenzilumab and anti-mouse GM-CSF antibody prevent CRS-induced weight loss compared with mice treated with CART19 cells and isotype-control antibodies; 3 mice per group, 2-way analysis of variance. Data are mean ± SEM. (B) Human GM-CSF was neutralized in patient-derived xenografts treated with lenzilumab and mouse GM-CSF–neutralizing antibody; 3 mice per group. Data are mean ± SEM. *P < .05, ***P < .001, Student t test. (C) Human cytokine/chemokine heat map (serum collected 11 days after CART19 cell injection) exhibits increases in cytokines and chemokines typical of CRS after CART19 cell treatment. GM-CSF neutralization results in a significant decrease in several cytokines and chemokines compared with mice treated with CART19 cells and isotype-control antibodies, including several myeloid-associated cytokines and chemokines, as indicated in the panel; 3 mice per group, serum from day 11 post–CART19 cell injection. *P < .05, **P < .01, ***P < .001, GM-CSF neutralizing antibody–treated mice vs isotype control–treated mice that received CAR-T cell therapy, Student t test. (D) Mouse cytokine/chemokine heat map (serum collected 11 days after CART19 cell injection) exhibits increases in mouse cytokines and chemokines typical of CRS after CART19 cell treatment. GM-CSF neutralization results in a significant decrease in several cytokines and chemokines compared with treatment with CART19 cells with control antibodies, including several myeloid-differentiating cytokines and chemokines, as indicated in the panel; 3 mice per group, serum from day 11 post–CART19 cell injection, *P < .05, GM-CSF neutralizing antibody–treated mice vs isotype control–treated mice that received CAR-T cell therapy, Student t test.

GM-CSF neutralization in vivo ameliorates NI after CART19 cell therapy in a xenograft model. (A-B) Gadolinium-enhanced T1-hyperintensity (cubic millimeters) MRI showed that GM-CSF neutralization helped to reduced brain inflammation, blood–brain barrier disruption, and possible edema compared with isotype control. (A) Representative images. (B) Three mice per group. Data are mean ± SD. *P < .05, **P < .01, 1-way analysis of variance. (C) Human CD3 T cells were present in the brain after treatment with CART19 cell therapy. GM-CSF neutralization resulted in a decreased raw average of CD3 infiltration in the brain (although not statistically significant), as assayed by flow cytometry in brain hemispheres; 3 mice per group. Data are mean ± SEM. *P < .05, Student t test. (D) CD11b+ bright macrophages were decreased in raw average (although not statistically significant) in the brains of mice receiving GM-CSF neutralization during CAR-T cell therapy compared with those receiving isotype-control treatment during CAR-T cell therapy, as assayed by flow cytometry in brain hemispheres; 3 mice per group. Data are mean ± SEM, Student t test.
GM-CSF neutralization in vivo ameliorates NI after CART19 cell therapy in a xenograft model. (A-B) Gadolinium-enhanced T1-hyperintensity (cubic millimeters) MRI showed that GM-CSF neutralization helped to reduced brain inflammation, blood–brain barrier disruption, and possible edema compared with isotype control. (A) Representative images. (B) Three mice per group. Data are mean ± SD. *P < .05, **P < .01, 1-way analysis of variance. (C) Human CD3 T cells were present in the brain after treatment with CART19 cell therapy. GM-CSF neutralization resulted in a decreased raw average of CD3 infiltration in the brain (although not statistically significant), as assayed by flow cytometry in brain hemispheres; 3 mice per group. Data are mean ± SEM. *P < .05, Student t test. (D) CD11b+ bright macrophages were decreased in raw average (although not statistically significant) in the brains of mice receiving GM-CSF neutralization during CAR-T cell therapy compared with those receiving isotype-control treatment during CAR-T cell therapy, as assayed by flow cytometry in brain hemispheres; 3 mice per group. Data are mean ± SEM, Student t test.
Patient-derived xenograft model for NI and CRS
In this model, conditioned NSG mice were engrafted with patient ALL blasts and monitored for engraftment for several weeks until they developed high disease burden (Figure 4A). When the level of CD19+ blasts in the peripheral blood was ≥10 per microliter, mice were randomized to receive different treatments, as indicated (Figure 4A). Treatment with CART19 cells (with control IgG antibodies or with GM-CSF–neutralizing antibodies) successfully eradicated the disease (Figure 4B). Within 4-6 days after treatment with CART19 cells, mice began to develop motor weakness, hunched bodies, and progressive weight loss, symptoms that are consistent with CRS and NI. This was associated with elevation of key serum cytokines and chemokines 4-11 days post–CART19 cell injection, similar to what is seen in human CRS after CAR-T cell therapy (including human GM-CSF, TNF-α, IFN-γ, IL-10, IL-12, IL-13, IL-2, IL-3, IP-10, MDC, MCP-1, MIP-1α and MIP-1β and mouse IL-6, GM-CSF, IL-4, IL-9, IP-10, MCP-1, and monokine induced by IFN-γ [MIG]). These mice treated with CART19 cells also developed NI, as indicated by brain magnetic resonance imaging (MRI) analyses revealing abnormal T1 enhancement, suggestive of blood–brain barrier disruption and possibly brain edema24 (Figure 4C), together with flow cytometric analysis of harvested brains revealing infiltration of human CART19 cells (Figure 4D). In addition, RNA sequencing analyses of brain sections harvested from mice that developed these signs of NI showed significant upregulation of genes regulating the T-cell receptor, cytokine receptors, T-cell immune activation, T-cell trafficking, and T-cell and myeloid cell differentiation (Figure 5; supplemental Table 1).
GM-CSF neutralization in vivo ameliorates CRS and NI after CART19 cell therapy in a xenograft model
Using the xenograft patient-derived model for NI and CRS shown in Figure 4A, we investigated the effect of GM-CSF neutralization on CART19 cell toxicities. To rule out the confounding effect of mouse GM-CSF, mice received CART19 cells in combination with 10 days of GM-CSF antibody therapy (10 mg/kg lenzilumab and 10 mg/kg anti-mouse GM-CSF neutralizing antibody) or isotype-control antibodies. GM-CSF–neutralizing antibody therapy reduced CRS-induced weight loss after CART19 cell therapy in a statistically significantly manner (Figure 6A). Cytokine and chemokine analysis 11 days after CART19 cell therapy showed that human GM-CSF was neutralized by the antibody (Figure 6B). In addition, GM-CSF neutralization resulted in a significant reduction in several human (IP-10, IL-3, IL-2, IL-1 receptor antagonist, IL-12p40, vascular endothelial growth factor, GM-CSF) (Figure 6C) and mouse (MIG, MCP-1, KC, IP-10) (Figure 6D) cytokines and chemokines. IP-10 (CXCL10) is produced by monocytes, among other cell types, and serves as a chemoattractant for numerous cell types, including monocytes, macrophages, and T cells. IL-3 plays a role in myeloid progenitor differentiation. IL-2 is a key T cell cytokine. IL-1 receptor antagonist inhibits IL-1. (IL-1 is produced by macrophages and is a family of critical inflammatory cytokines.) IL-12p40 is a subunit of IL-12, which is produced by macrophages, among other cell types, and can encourage T helper 1 cell differentiation. Vascular endothelial growth factor encourages blood vessel formation. MIG (CXCL9) is a T-cell chemoattractant. MCP-1 (CCL2) attracts monocytes, T cells, and dendritic cells. KC (CXCL1) is produced by macrophages, among other cell types, and attracts myeloid cells, such as neutrophils. There was also a nonstatistically significant reduction in several other human and mouse cytokines and chemokines after GM-CSF neutralization. This suggests that GM-CSF plays a role in the downstream activity of several cytokines and chemokines that are instrumental in the cascade that results in CRS and NI.
Brain MRIs 5 days after CART19 cell treatment showed that GM-CSF neutralization reduced T1 enhancement, as a measure of brain inflammation, blood–brain barrier disruption, and possibly edema, compared with CART19 cells plus control antibodies. The MRI images after GM-CSF neutralization (with lenzilumab and anti-mouse GM-CSF antibody) were similar to control scans, suggesting that GM-CSF neutralization effectively helped to abrogate the NI associated with CART19 cell therapy (Figure 7A-B). Using human ALL blasts and human CART19 cells in this patient-derived xenograft model, GM-CSF neutralization after CART19 cells reduced NI by 75% compared with CART19 cells plus isotype controls (Figure 7B). This is a significant finding, and it has been demonstrated for the first time in vivo that the NI caused by CART19 cells can be effectively abrogated. Human CD3 T cells were present in the brain after CART19 cell therapy, as assayed by flow cytometry, and with GM-CSF neutralization there was a difference in raw average with a reduction in brain CD3 T cells; however, it did not meet statistical significance (Figures 4D, 7C; supplemental Figure 6). Finally, a difference in raw average (although this did not reach statistical significance), with a reduction in CD11b+ bright macrophages, was observed in the brains of mice receiving GM-CSF neutralization during CAR-T cell therapy compared with those receiving isotype control during CAR-T therapy (Figure 7D), possibly implicating that GM-CSF neutralization helps to reduce macrophages within the brain.
Discussion
Our results provide the first proof of concept that neutralization of GM-CSF abrogates toxicities after CAR-T cell therapy and may enhance their therapeutic activity. Specifically, we have shown that GM-CSF neutralization, in combination with CART19 cell therapy, prevents the development of CRS and significantly reduces the severity of NI in a xenograft model using human ALL blasts and human CART19 cells. GM-CSF neutralization resulted in a reduction in chemokines associated with myeloid trafficking, such as IP-10, MCP-1, KC, and other inflammatory cytokines and chemokines, and is associated with decreased raw averages (although not statistically significant) of T cell infiltration and myeloid cell activation in the brain. Intriguingly, our experiments also suggest that GM-CSF inhibition enhances CART19 cell proliferation, antitumor activity, and overall survival in vivo. Based on the results reported here, GM-CSF neutralization can be viewed as a potential next-generation strategy to enable routine CAR-T cellular immunotherapy.
In our studies, GM-CSF neutralization with lenzilumab did not impair any CART19 cell effector functions in vitro. In 2 xenograft models (NALM6 xenografts and patient-derived xenografts), CART19 cells combined with lenzilumab effectively eradicated the tumor, despite GM-CSF neutralization, and significantly improved leukemic disease control 35 days posttreatment, whereas CART19 cells plus isotype control could not maintain disease control after 35 days. Lastly, GM-CSFk/o CART19 cells exhibited potent effector functions in vitro and demonstrated significantly improved overall survival compared with CART19 cells in vivo.
Our CRS and NI model is a unique and relevant ALL patient–derived xenograft model for the development of therapies for toxicities after human CAR-T cell therapy. In our model described here, the time interval between CAR-T cell infusion and the onset of symptoms, brain MRI changes, cytokine and chemokine elevation, and infiltration of effector cells into the CNS are all similar to what is reported in patients who develop toxicities after CART19 cell therapy. Mice developed symptoms of CRS and NI (weight loss, decline in motor function, and hunched bodies). Changes in brain MRI were detected 4 to 6 days after infusion of CART19 cells. Brain MRI T1 uptake is suggestive of blood–brain barrier disruption and, possibly, brain edema24 and is comparable to changes noted on human brain MRIs in cases of severe neurotoxicity, as described by Gust et al.8 Interestingly, Gust et al further describe that blood–brain barrier permeability prevented protection of the CSF from systemic cytokines, which induced vascular pericyte stress and secretion of endothelium-activating cytokines, and patients showed evidence of endothelial activation.8 In our CRS/NI model, GM-CSF was found to be neutralized in the serum of mice receiving CART19 cell therapy with GM-CSF neutralizing antibodies compared with CART19 cells and isotype-control antibodies. Thus, T cells within the mouse brains themselves might produce GM-CSF, and serum GM-CSF, among other cytokines and chemokines, was possibly able to reach the CSF. In addition, endothelium cells are able to produce GM-CSF, which may result in a cycle of exacerbation.25 NI was associated with infiltration of T cells and activation of myeloid cells in the CNS, similar to CSF changes in patients with CAR-T–induced neurotoxicity,8,12,13 as well as in nonhuman primate models.26 Our model is similar to previously reported patient-derived xenograft models in which CRS developed after CAR-T cell therapy.27,28 A recent report suggested that blockade of IL-1 prevents NI through the depletion of myeloid cells.29 However, the development of NI in that model was delayed and related to meningeal thickening, unlike what was observed in our model and in patients receiving CART19 therapy. Therefore, we propose our model as a reliable way to investigate novel interventions for the prevention and treatment of CRS and neurotoxicity after CART19 cell therapy. Our results show that GM-CSF neutralization results in a reduction in key myeloid and several inflammatory cytokines and chemokines, suggesting that GM-CSF is a critical cytokine in the downstream activation of several cytokines and chemokines, blockade contributes to a decrease in raw averages in myeloid cell and T-cell infiltration in the brain/CNS (although statistical significance was not reached), and blockade helps to reduce NI of apparent neurotoxicities.
Interestingly, we observed an exponential increase in CART19 cell proliferation, enhanced antitumor activity, and improved overall survival with GM-CSF blockade. For example, CART19 cell antigen–specific proliferation in the presence of monocytes increased in vitro after GM-CSF neutralization. Moreover, in ALL patient–derived xenografts, CART19 cells resulted in a more durable disease control when combined with lenzilumab. In addition, we found that GM-CSFk/o CAR-T cells were more effective in controlling leukemia in NALM6 xenografts and demonstrated improved overall survival. Although the mechanisms for enhanced CAR-T effector functions after GM-CSF depletion are unclear and beyond the scope of this article, our results are consistent with previous reports indicating that monocytes impair T cell expansion ex vivo and that M2-polarized macrophages inhibit CART19 cell antigen–specific proliferation.30 This is an important finding because, across CAR-T cell clinical trials, improved CAR-T cell proliferation was consistently associated with improved efficacy and response (ie, overall and complete response rates).3,5,7,31
It is known that activated T cells produce GM-CSF.25 T cells do not possess all of the subunits for the GM-CSF receptor; therefore, under ordinary circumstances, GM-CSF usually does not feedback on T cells directly, although it can, under some circumstances, at very high levels.25 Instead, this GM-CSF affects the behaviors of numerous other cell types, including macrophages and dendritic cells.25 The subsequent activation of these cells results in actions that work to stimulate T cells, such as cytokine production and antigen presentation.25 In turn, T-cell stimulation can further drive production of GM-CSF and other cytokines to act on the other cell types, like macrophages and dendritic cells, which drives the cycle.25 In CAR-T cell therapy, it is likely that the large number of activated T cells produced over a very short time pushes this cycle to an extreme situation. Our results suggest that blocking GM-CSF helps to prevent this immune overstimulation without impairing T-cell functions, actually enhancing them. The exact mechanisms for enhanced CAR-T cell effector functions after GM-CSF blockade are unclear and beyond the scope of this article. Appropriate mechanistic studies will be performed and reported in a follow-up publication.
Finally, our results additionally suggest that the development of GM-CSFk/o CART19 cells may represent a novel way to partially control GM-CSF production that can be incorporated into current CAR-T cell manufacturing. Our results indicate that these cells function normally and could represent an independent therapeutic approach to enhance the therapeutic window after CAR-T cell therapy. With lenzilumab, we have a clinical-stage therapeutic solution to neutralize GM-CSF, abrogate CRS and NI of apparent neurotoxicities, and potentially improve CAR-T cell function.
This study represents a significant advance in understanding and preventing toxicities after CAR-T cell therapy. Our results strongly suggest that modulating myeloid cell behavior through GM-CSF blockade helps to control CAR-T cell–mediated toxicities and reduce their immunosuppressive features to improve leukemic control. These studies illuminate a novel approach to abrogate NI of apparent neurotoxicities and CRS through GM-CSF neutralization that also potentially enhances CAR-T cell functions. Based on these results, a phase 2 trial of the combination of CART19 cells and lenzilumab is planned.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from Humanigen (S.S.K.), K12CA090628 (S.S.K.), the National Comprehensive Cancer Network (S.S.K.), the Mayo Clinic Center for Individualized Medicine (S.S.K.), National Institutes of Health, National Institute of Neurological Disorders and Stroke grant R01 NS103212 (A.J.J.), the Mayo Clinic-Koch Institute Collaboration (A.J.J.), and the Mayo Clinic Medical Scientist Training Program Robert L. Howell Physician-Scientist Scholarship (R.M.S.).
Authorship
Contribution: R.M.S. and S.S.K. designed experiments and wrote the manuscript; R.M.S., S.S.K., R.S., and M.J.C. analyzed data; R.M.S., R.S., M.J.C., N.Y., and C.L.F. performed in vitro and in vivo experiments; M.J.H. performed in vivo experiments; R.H.K., F.J., and K.A. performed MRI imaging and analyzed the data; D.K.W., M.H., K.J.S., and W.K.N. assisted with experiments; A.J.J., N.E.K., K.E.H., L.R.P., M.M.P., C.D., T.S., D.C., and O.A. provided guidance and expertise in their respective areas of study; S.S.K. supervised the study; and all authors provided input and edited and approved the final version of the manuscript.
Conflict-of-interest disclosure: S.S.K. is an inventor on patents in the field of CAR-T cell therapy that are licensed to Novartis (under an agreement among the Mayo Clinic, the University of Pennsylvania, and Novartis). O.A., T.S., D.C., and C.D. are employed by Humanigen. R.M.S., R.S., M.J.C., and S.S.K. are inventors on patents related to this work. The remaining authors declare no competing financial interests.
Correspondence: Saad S. Kenderian, Mayo 10-30E, 200 First St Southwest, Rochester, MN 55905; e-mail: kenderian.saad@mayo.edu.
