

Abstract
Hypomethylating agents (HMA) azacitidine and decitabine are standard of care for myelodysplastic syndrome (MDS). Response to these agents occurs in ∼50% of treated patients, and duration of response, although variable, is transient. Prediction of response to HMAs is possible with clinical and molecular parameters, but alternative approved treatments are not available, and in the case of HMA failure, there are no standard therapeutic opportunities. It is important to develop a reasoned choice of therapy after HMA failure. This choice should be based on evaluation of type of resistance (primary vs secondary, progression of disease [acute leukemia or higher risk MDS] vs absence of hematological improvement) as well as on molecular and cytogenetic characteristics reassessed at the moment of HMA failure. Rescue strategies may include stem-cell transplantation, which remains the only curative option, and chemotherapy, both of which are feasible in only a minority of cases, and experimental agents. Patients experiencing HMA failure should be recruited to clinical experimental trials as often as possible. Several novel agents with different mechanisms of action are currently being tested in this setting. Drugs targeting molecular alterations (IDH2 mutations, spliceosome gene mutations) or altered signaling pathways (BCL2 inhibitors) seem to be the most promising.
Introduction
For more than a decade, hypomethylating agents (HMAs) azacitidine and decitabine have been considered standard of care for myelodysplastic syndrome (MDS).1,2 Despite their low toxicity and ability to induce hematological improvement (HI) and prolongation of survival, even in elderly patients, HMAs are not curative without hematopoietic stem-cell transplantation (HSCT),3,4 and despite responses, they do not eradicate neoplastic clones.3,5 The enthusiasm generated by their large applicability and activity has been followed by a wave of critical appraisal stemming from evidence accumulated in the years of clinical use of these drugs. In practice, the proportion of those with MDS who respond and maintain response for a substantial period comprises fewer than half of treated patients. Published results of HMA-treated patients in controlled trials indicate better outcomes compared with real-life data. This inconsistency may be due to differences in adherence to dose, schedule, and minimum number of cycles, as well as to the management of patients with severe comorbidities.6-8 Overall, it is clear that the HMA effect is transient, with responses maintained for 6 to 24 months.4,8 Survival of those with refractory/relapsed disease is extremely short, for both International Prognostic Scoring System (IPSS) lower-risk and high-risk MDS patients.9-11 In Europe, the European Medicines Agency approved HMA therapy for IPSS higher-risk MDS patients, whereas in the United States and many other countries, HMAs are prescribed for patients with all types of MDS. This particularity differentiates European patients with HMA-resistant/relapsed disease, all of whom have bad prognoses, whereas elsewhere, these patients may still be considered lower risk and have diverse therapy options.
Optimal management of therapy with HMAs
How do I manage HMA failure in absence of a clinical trial?
Patient 1: high-risk MDS
A 70-year-old woman with IPSS intermediate 2–risk MDS with multilineage dysplasia (trisomy 8 and 18% marrow blasts, SRSF2 and ASXL1 mutations) received 8 cycles of azacitidine at standard dose and schedule, combined with eltrombopag, in a phase 3 experimental trial (registered at www.clinicaltrials.gov as #NCT02158936), with resulting neutropenia, without significant increase in platelets, and with only slight decrease in marrow blasts. The patient had no comorbidities, and her general condition was optimal. At routine control during follow-up, peripheral blood blasts were detected (4%), and bone marrow (BM) aspirate confirmed progression (30% BM blasts). No experimental options were available at that time for MDS that has progressed to acute myeloid leukemia (AML) after HMAs. The patient had a Sorror score <3 and did not find an HLA-matched donor from the registry. Two months later, she developed severe thrombocytopenia and accepted a haploidentical transplant from her daughter in an experimental approach. She did not receive preemptive chemotherapy, but a conditioning regimen of fludarabine, busulfan, and thiotepa before transplantation was administered. The procedure was successful, with only grade 1 acute graft-versus-host disease and no chronic graft-versus-host disease with mycophenolate mofetil, cyclosporine, and cyclophosphamide. Her counts normalized within 2 months, marrow blasts were absent, and her Karnofsky status was 90%. This patient had progressed to AML after HMA failure and underwent transplantation when she had active disease in advanced age. Haploidentical transplantation in a clinical study setting allowed her to survive 16 months with optimal quality of life. It is indeed worthwhile discussing transplantation options in fit patients with no other therapeutic opportunities.
Patient 2: low-risk MDS
An 80-year-old woman with a diagnosis of MDS with multilineage dysplasia, trisomy 8, and 4% BM blasts (IPSS risk, intermediate-1; IPSS revised [IPSS-R] risk, intermediate) was in need of sporadic red blood cell (RBC) transfusions. She was determined to be erythropoietic stimulating agent (ESA) refractory after <2 months of therapy, and she received azacitidine at a flat dose of 100 mg/m2 per 7 days monthly subcutaneously for 3 months without any HI. She came to our center and received 9 months of standard-dose azacitidine, and after 12 months of stable disease, she had a further decrease in hemoglobin (Hb) and absolute neutrophil count levels. Suspecting progression, BM aspiration and biopsy were performed, and marrow was found to be highly hypocellular (<20% cellularity) but without increased blasts; cytogenetic analysis revealed trisomy 8 and the novel detection of del5q (20% metaphases). She was administered erythropoietin subcutaneously, achieving transient normalization of Hb levels, after which she again became RBC transfusion dependent. Treatment with lenalidomide was started at 10 mg per day. After 6 months, her counts were: Hb, 14.4 g/dL; mean corpuscular volume, 98.4 fL; platelets, 160 × 109/L; white blood cell (WBC) count, 4.0 × 109/L; and absolute neutrophil count, 2.1 × 109/L. Kidney function slightly decreased, but otherwise, she had excellent general clinical conditions. Anemic low-risk MDS patients may need several weeks to respond to ESAs; treatment should be prolonged at least 8 weeks before transitioning to HMA therapy. A complete revaluation of the patient at HMA failure with marrow aspirate, biopsy, and reassessment of cytogenetics is mandatory, because it may provide useful indications for sequential treatments. Long-term treatment with HMAs may rarely induce marrow hypocellularity. Dysplastic clones may arise, allowing targeted therapy opportunities.
Optimization of management of HMAs to avoid treatment failure, with correct drug doses, schedules, and timing of evaluations of response, and subsequent clear definition of resistance12 are fundamental. In case 2, failure of therapy was declared too early and after reduced HMA dosing.
Timing of evaluation
Conclusive assessment of response should be performed in MDS patients treated with standard dose of decitabine (20 mg/m2 per day for 5 days at 4-week intervals) or azacitidine (75 mg/m2 per day for 7 days at 4-week intervals) for at least 6 cycles. Earlier evaluations may fail to detect responses, achieved in a majority of cases between cycles 4 and 6, although later responders may be encountered. Premature interruption of therapy leads to rapid loss of response, and rechallenge with HMAs is not usually effective. Simple follow-up of patients during HMA therapy is by monitoring peripheral blood counts, reappearance of severe cytopenias, and/or appearance of blasts. BM evaluation can be performed at appropriate time intervals (ie, every ≥6 months, earlier in the suspect of progression).12
Dosing and schedules
Several alternative dose and schedule regimens have been evaluated in retrospective real-life studies, but formal demonstration of clinical advantage was shown with the doses and schedules mentioned in “Timing of evaluation.” Large randomized trials comparing alternative dosages are lacking. A recent meta-analysis indicated the inconsistency of the results with alternative doses and the equivalence of schedules of 5-2-2 days with the standard 7 days in terms of response.13
Reinforcing HMA therapy
Empirical addition of other agents to HMAs in front-line therapy, like histone deacetylase inhibitors,14-18 lenalidomide,16,19 or even cytotoxic agents,20 has not substantially improved outcomes. Randomized phase 3 combination studies combining HMAs with various agents (ie, combination of azacitidine with pevonedistat [#NCT03268954] or venetoclax [#NCT02942290]) are presently ongoing for first-line therapy of MDS.
Definition of HMA resistance
Even when HMA therapy is conducted with correct schedules and for a sufficient number of cycles, failure may occur in different scenarios.
Primary resistance
Primary resistance occurs when, while receiving therapy without experiencing HI at any time, the patient progresses to overt acute myeloid leukemia (>20% BM blasts); the patient progresses to higher-risk MDS; even after 4 to 6 cycles, the patient has stable disease without any of the following: HI, complete remission (CR), marrow CR (mCR), or partial remission (PR), according to International Working Group criteria21 ; or the patient develops hypoplastic marrow and pancytopenia.
Secondary resistance
Secondary resistance occurs when, after initial response (CR, mCR, PR, HI) has been maintained for any number of cycles and without therapy interruption or delays exceeding 5 weeks between cycles, the treated patient has any of the primary resistance conditions. These situations may be encountered in both higher-risk and lower-risk MDS patients receiving azacitidine or decitabine.
Although application of International Working Group criteria21 outside clinical trials is cumbersome, strict adherence may present difficulties even in controlled studies, and a revision of these criteria has been proposed.22
We have to solve the riddle of how to treat MDS patients who do not respond to HMAs (primary resistant) or whose response does not last (secondary resistant), but at present we approach these 2 types of failure with the same experimental agents (Figure 1).
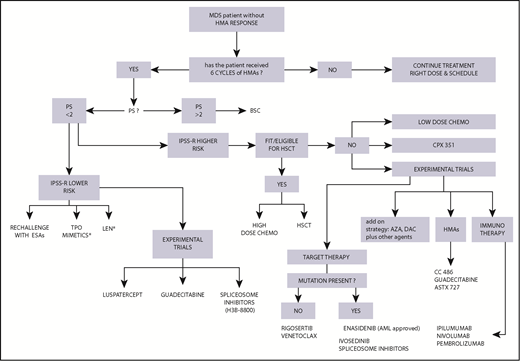
Algorithm for treatment choice in MDS patients after failure of HMA therapy. Reasoned choice of second- and third-line treatment options after HMA therapy based on clinical and biological characteristics of the patient. In this setting, experimental trials are recommended; the experimental agents indicated in the figure are those evaluated in the highest number of cases. *Off-label use; on-label use of lenalidomide (LEN) only in del5q; thrombopoietin (TPO) mimetics in experimental trials. AZA, azacitidine; chemo, chemotherapy; DAC, decitabine; PS, performance status.
Algorithm for treatment choice in MDS patients after failure of HMA therapy. Reasoned choice of second- and third-line treatment options after HMA therapy based on clinical and biological characteristics of the patient. In this setting, experimental trials are recommended; the experimental agents indicated in the figure are those evaluated in the highest number of cases. *Off-label use; on-label use of lenalidomide (LEN) only in del5q; thrombopoietin (TPO) mimetics in experimental trials. AZA, azacitidine; chemo, chemotherapy; DAC, decitabine; PS, performance status.
Indeed, the most relevant obstacle to developing target treatment is the lack of knowledge of the mechanisms of resistance to HMAs, but supposing hypomethylation is the main mechanism of action of these agents, attention has been focused on epigenetic modulation.
DNA methylation
The lack of correspondence between the pattern of DNA hypo- or hypermethylation and response to HMAs demonstrated in numerous studies23-28 underlines the limited knowledge of chromatin regulation and HMA mechanisms of action.29 We showed that decitabine response is predictable in chronic myelomonocytic leukemia by baseline differentially methylated regions in primary resistant vs sensitive patients.28 Differentially methylated regions were not preferentially localized in promoters but rather in enhancers and intergenic regions and corresponded to high production of cytokines like CXCL4 and CXCL7.28 While we wait confirmation, it remains an open question whether epigenetic alterations are different in primary vs secondary HMA resistance.
Recently, primary resistance to azacitidine was linked to hematopoietic progenitor cell cycle quiescence,30 mediated by integrin a5 signaling. This would provide a target, because responsiveness to azacitidine was restored by exposure to an anti–integrin 5a antibody. HMAs, even when clinically effective, failed to eradicate clonal hematopoiesis, but they restored functional hematopoiesis from progenitors with a lower mutational burden by altering the subclonal architecture of the hematopoietic progenitor compartment.30
The involvement of nucleoside activating/deactivating enzymes31 and membrane transporters32 was shown to influence HMA response by putative modulation of nucleoside-triphosphate uptake into DNA.32 These observations have recently been challenged,33 but they nevertheless prompted development of an oral formulation of decitabine combined with the cytidine deaminase inhibitor cedazuridine ASTX727 (#NCT02103478).34
Response to HMAs and somatic mutations
Many acquired somatic mutations in MDS affect epigenetic mechanisms and are present in ∼90% of cases; they could drive MDS pathophysiology and HMA response/resistance.35 TET2 mutations were correlated with DNA hypermethylation35,36 and HMA response, but not with overall survival (OS).36,37 DNMT3A mutations, accompanied by a hypomethylated profile,38 also correlated only with HMA response. Therefore, quantitation of DNA methylation per se cannot be the reason why these mutations can modulate HMA sensitivity. The mutation of ASXL1 predicted poor outcome in terms of response38 and OS after HMAs38 ; similarly, TP53 mutations correlated with dismal outcome.23 Recently, 10-day treatment with decitabine was demonstrated to induce response in all TP53-mutated cases,39 possibly indicating a role of the disruption of TP53 function in HMA sensitivity.40
The presence of specific somatic mutations does not yet influence the decision to treat with HMAs. In general, it suggests more or less aggressive strategies (eg, earlier timing of HSCT for eligible patients carrying numerous mutations) and drives second-choice therapies when genes like IDH1/IDH241,42 or spliceosome components42 are mutated, for which specific inhibitors42,43 are available. The patient presented in case 1 had an ASXL1 mutation, among others, and transplantation seemed the best option.
Prediction of response
It would be extremely important to predict response and survival after HMAs to adjust personalized rescue therapy. A method was recently proposed to predict outcomes of patients after HMA failure: the post-HMA model. It includes 6 variables: age, PS, complex cytogenetics (>3 abnormalities), marrow blast >20%, platelet count, and red cell transfusion dependency.44 This model, validated but not commonly applied,45,46 separates MDS patients evaluated after HMA failure into 2 risk categories: lower risk, with a median OS of 11.0 months; and higher risk, with median OS of 4.5 months.
Management of HMA failures
The options for therapy after HMA failure are scarce, and although experimental clinical trials are the recommended measure, when not available, supportive care, ESAs, HSCT, high- or low-dose chemotherapy, lenalidomide, and alternative sequencing of HMAs can be employed.
Decision making at the time of failure must include, after complete clinical examination, a thorough discussion with the patient and caregivers. Patient personal choices, attitudes, and inclination are fundamental. Best supportive care, including growth factors like ESAs and possible consideration of thrombopoietic agents, is on option the treating physician cannot exclude a priori without analyzing the situation together with the patient. In some cases, it may represent the option ensuring the best quality of life or duration of survival, especially in frail patients or those with several comorbidities. Although elderly, the patient in case 2 was extremely active and insisted on being treated.
HSCT
MDS patients are generally age ≥70 years, frequently present with comorbidities, and may be extremely frail. Therefore, only a small minority of MDS patients are eligible for transplantation, although this number is increasing because of wider choice of donor (sibling, unrelated, cord blood, haploidentical) and different conditioning regimens that render the procedure less risky.47 After HMA failure, as is clear from case 1, HSCT is feasible and results in prolonged survival compared with other treatments.10 There are at present no prospective studies evaluating the outcome of MDS patients undergoing transplantation after HMA failure, but a retrospective analysis of this subpopulation indicated relapse-free survival at 3 years was 23.8%, an encouraging result compared with other treatments.48 Pretransplantation selection of MDS patients after HMA failure could improve HSCT outcome. Patients with TP53 mutations undergoing HSCT have shorter survival and earlier relapse, irrespective of conditioning regimen,49,50 and the advantages and risks of transplantation should be well weighed.51,39
High-dose chemotherapy
Intensive AML-like treatment of MDS patients after HMA failure was reported, with 8.9-month survival.10 Recently, in a large number of MDS patients (n = 307), of whom 31% were IPSS lower risk, treated after HMA failure with cytosine arabinoside plus anthracycline (7+3), intermediate- to high-dose cytosine arabinoside, or nucleoside analogs, median OS was 10.8 months and overall response rate (ORR) was 41%.52 Negative prognostic factors were adverse cytogenetics, age ≥65 years, and use of intermediate-dose cytosine arabinoside.52 Standard-dose clofarabine used after HMA failure in patients with advanced MDS showed excessive toxicity.53
Low-dose chemotherapy
The use of subcutaneous low-dose cytarabine after HMA failure does not have any advantage over supportive care,10 with absence of response and OS of 7.4 months. Combination of cytarabine with experimental agents has not shown encouraging results. In contrast, the addition of low-dose clofarabine to low-dose cytarabine54 to treat elderly patients with MDS after HMA failure yielded a 44% ORR and OS of 10 months.
Lenalidomide
In lower-risk MDS patients who are resistant/refractory to ESAs and HMA treatment, lenalidomide is an available option. Administered after azacitidine, lenalidomide is well tolerated, but in non–del5q MDS, it induces a limited 12% erythroid improvement55 and an OS of 87 months. In high-risk MDS after HMA failure, treatment with standard- (15 mg) or high-dose (50 mg) lenalidomide resulted in scarce/absent clinical activity and extreme toxicity.56 However, there are reports of higher response rates (40%) in HMA-refractory MDS patients, with CR in those carrying del5q.57 High doses of lenalidomide induced mCR in 33% and HI in 8% of HMA-refractory patients.58 The patient in case 2 responded and had a del5q clone; cytopenia occurred, but blasts were <5% after azacitidine.
Sequential use of HMAs
In the absence of experimental trials and for unfit or very elderly patients for whom HMAs have failed, an HMA alternative to the HMA that failed has been administered. Although switching may be suggested when patients experience intolerance, results of this strategy in truly resistant cases are far from successful, as in case 4. The scattered data published derive from small retrospective studies lacking precise definitions of HMA resistance. Response rates to decitabine after azacitidine are <30%,59-61 and only 1 study reported a response rate of 40% for patients who received azacitidine after first-line decitabine.60 Because of the slightly different mechanisms of action of the 2 agents, switching may be partially justified, but recent evidence33 suggests that azacitidine is clinically active only when incorporated into DNA, with a mechanism overlapping that of decitabine. In any case, the prolonged duration of HMA treatment seems to account for the increased responses observed when the second HMA is sequentially introduced.
Experimental agents and combinatory experimental trials
There are many clinical studies ongoing testing different agents for treatment of MDS after HMA failure. Because of the extreme need for novel treatments for these patients, several investigatory trials are empirical, but the most promising studies are based on the presence and so-called druggability of specific molecular targets.
How do I select a clinical trial after HMA failure?
Patient 3
A 74-year-old man with MDS excess blasts 2 and IPSS-R high risk was treated with azacitidine for 24 months with optimal hematological and cytogenetic responses, but he subsequently relapsed. BM aspiration indicated 20% blasts with normal karyotype. This secondary resistant patient was referred to our center, and the possibility of recruitment to an experimental study was discussed with him and his family. General clinical conditions were acceptable (PS 2). After ruling out the presence of somatic mutations and confirming the patient could not be enrolled in clinical trials with target drugs, participation was proposed in a phase 3 trial randomizing patients to guadecitabine (#NCT02907359) vs low-dose cytarabine. The problem of randomization was again discussed to clarify the procedure. The patient was randomized to the guadecitabine arm and initiated therapy a few days later at 60 mg/m2 subcutaneously for 5 days in a 28-day cycle. Treatment is ongoing and well tolerated, with some myelosuppressive effects and no general toxicity.
Novel HMAs
Given the fact that HMAs were the first type of agents able to improve outcome in MDS, novel molecules with hypomethylating activity have been synthesized. Guadecitabine (SGI-110) is a dinucleotide that couples decitabine to deoxyguanosine and is resistant to deamination by cytosine deaminase, with prolonged in vivo exposure and clinical activity in de novo AML and high-risk MDS patients.62 Preliminary phase 2 data indicated some efficacy after HMA failure,63 with a toxicity profile similar to that of standard HMAs. A phase 3 randomized trial is currently ongoing for MDS after HMA failure, comparing guadecitabine with treatment of choice (#NCT02907359).
With the same idea of prolonging the stability of decitabine, the novel oral compound ASTX727 combines the cytidine deaminase inhibitor cedazuridine with decitabine. This agent has been administered in a recent phase 1 study to MDS patients after HMA failure, resulting in a 32% ORR.34 After the pharmacokinetic study of ASTX727,34 a phase 3 randomized study will compare IV decitabine bioavailability and clinical activity with those of ASTX727 (#NCT03306264). Quite clearly, oral therapy would significantly improve quality of life in elderly MDS patients. A phase 2 clinical trial with cc-486 monotherapy in HMA-refractory MDS is ongoing. This oral agent should provoke longer exposure to azacitidine than the subcutaneous analog (#NCT02281084).
Patient 4
A 70-year-old woman was referred after having lost response (PR) to 12 cycles of standard-dose azacitidine administered for MDS excess blasts 2, secondary to chemotherapy for lymphoma. Because of her deteriorating condition (PS 2) and the presence of peripheral blood blasts (20%), she was treated with 3 cycles of decitabine at 20 mg/m2 per day for 5 days, without any improvement, and subsequently with 1 cycle of low-dose cytarabine, which was stopped for a Clostridium difficilis infection. We evaluated karyotype and presence of somatic mutations, but in the meantime, she rapidly developed overt leukemia with severe leukocytosis: WBC, 140 × 109/L, with 63% peripheral blood blasts; Hb, 6.8 g/dL; and platelets, 39 × 109/L. Therapy with hydroxiurea at 2000 mg per day was introduced to decrease leukocytosis. Mutational analysis indicated IDH2 mutation R172K. She was screened for the AML study protocol (#NCT02577406) comparing as rescue therapy enasidenib at 100 mg per day on a 28-day cycle with physician choice of therapy (low-dose cytarabine, high-dose chemotherapy, or best supportive care). At randomization, she was assigned to the enasidenib treatment arm. She started treatment in association with hydroxiurea for 2 weeks and had recurrent bacterial infections for which received IV antibiotics. Treatment with enasidenib was never stopped, and she experienced in 8 weeks a progressive normalization of WBCs, decrease of blasts to 4% in peripheral blood, and normalization of platelets to 206 × 109/L. She had no signs of differentiation syndrome. She still received sporadic RBC transfusions (Hb levels of ∼9 g/dL), but at BM aspiration (cycle 3), blasts were 13%, whereas a majority of myeloid cells, although dysplastic, had the morphology of band cells and mature granulocytes.
Full reevaluation (including somatic mutation analysis) of patients at the time of HMA failure is worthwhile, because it may open new avenues of treatment with target agents.
Targeted therapies
IDH2 and IDH1 inhibitors
Enasidenib (formerly AG-221) is an orally available, selective, potent inhibitor of the mutated IDH2 protein, a mutation found in <10% of MDS cases.41 Its differentiating activity has been shown in MDS,64 with ORRs of ∼40% to 50% in a majority of pretreated patients. The drug was recently approved by the US Food and Drug Administration42 for relapsed AML harboring an IDH2 mutation. The phase 3 study (#NCT02577406) in which the patient in case 4 was enrolled is recruiting AML patients and high-risk MDS patients who progressed to AML while receiving therapy with HMAs. The effects of IDH1 inhibitors like ivosidenib are not yet evaluable, because of the limited number of MDS patients treated. Even if the MDS population with IDH1/IDH2 mutations is limited,41 the efficacy of these inhibitors produces high response rates and imposes molecular evaluation in cases of HMA failure and patients with post-MDS AML. Further investigation of enasidenib activity in MDS is ongoing in front-line therapy for HMA-naïve high-risk MDS in combination with azacitidine (#NCT03383575).
Spliceosome inhibitory agents
Various spliceosome genes are frequently mutated in MDS.43 Because of the good correlation between genotype/phenotype and prognostic significance, these altered genes constitute an ideal target for specific agents. Toxic consequences of inhibition of RNA splicing in the different tissues (mainly ocular toxicity) were clear from early studies of E710765 but are not present in the ongoing phase 1 study of H3B-8800 (#NCT02841540),66 an orally bioavailable modulator of the SF3B complex administered to patients experiencing HMA failure and those with pretreated AML and MDS. H3B-8800 binds mutant and wild-type SF3B complexes but exerts its cytotoxic activity on mutant cells, where inhibition of the aberrant spliceosome machinery leads to cell death.
Quite recently, 2 studies demonstrated high clinical activity of the TGF β pathway inhibitors luspatercept67 and sotatercept68 in pretreated IPSS lower-risk MDS refractory to ESAs and HMAs.68 In the study by Komrokji et al68 (carried out mainly in the United States, where HMAs are approved for lower-risk MDS), 48.6% of anemic patients had experienced HMA failure, and 58.3% of them achieved erythroid HI with sotatercept therapy. This finding is of particular interest, because treatment with sotatercept (and most probably with luspatercept) may rescue HMA-pretreated severely anemic lower-risk MDS patients who otherwise would have few treatment opportunities and dismal outcomes.
BCL2 inhibition
Venetoclax (ABT199) is an oral BCL2 inhibitor licensed for treatment of chronic lymphocytic leukemia. This agent demonstrated apoptotic activity in vitro in progenitor cells from higher-risk MDS patients,69 and it has been employed with success in combination with low-dose chemotherapy or HMAs for patients with relapsed AML and for a small number of MDS patients.70 In a heavily pretreated group of patients (>2 salvage therapies and 77% prior HMA therapy), the ORR was 21%. Venetoclax in combination with azacitidine or decitabine has shown striking activity in elderly patients with untreated AML (CR, 61%),71 and these results have prompted additional studies in MDS patients after HMA failure, including an ongoing phase 2 clinical trial investigating the ORR of venetoclax at 400 mg as a single drug or in combination with 10-day decitabine (#NCT03404193). Another study combining azacitidine with venetoclax is recruiting MDS patients after HMA failure (#NCT02966782). Of note, the addition of the BCL2 inhibitor to azacitidine yielded quite important myelosuppression.
Multikinase inhibition
Rigosertib (previously ON-01910.Na) blocks the activation of polo-like kinase, Akt, and phosphatidylinositol 3-kinase pathways. This drug was tested in several studies in refractory/relapsed MDS and showed ability to reduce marrow blasts.72 A phase 3 trial comparing rigosertib administered IV as continuous infusion with best supportive care for MDS patients intolerant, refractory, or relapsed after HMAs failed to achieve its primary objective. However, it demonstrated an advantage in terms of OS for patients with primary resistance to HMAs (OS, 8.5 vs 4.7 months with BSC), patients age <80 years, and patients who had not received >9 cycles of HMAs.73 An oral formulation of rigosertib is now under evaluation in combination with azacitidine.74
Immunotherapy
The immune checkpoint molecules programmed death 1 (PD-1)/PD-1 ligand (PD-L1) and CTLA-4 modulate T-cell activation and antitumor immune surveillance in solid tumors as well as in myeloid neoplasms. PD-1 and CTLA-4 are overexpressed in MDS and more so after HMA failure.75 For this reason, several clinical studies have been designed to evaluate immune checkpoint inhibitory antibodies after HMA failure: durvalumab (anti–PD-L1; #NCT02775903), ipilimumab (anti–CTLA-4),75,76 nivolumab,75 atezolizumab (anti–PD-L1),77 and pembrolizumab (anti–PD-1).78 Nivolumab, pembrolizumab, and atezolizumab are currently approved in solid tumors. Azacitidine and decitabine upregulate the expression of immune checkpoint molecules, and these antibodies were tested in MDS as monotherapies and in association with HMAs. Clinical trials in MDS are ongoing and have shown signs of activity, but final data are not yet available. A phase 1 study of durvalumab plus azacitidine in untreated high-risk MDS has been put on hold (#NCT02775903), and there are no studies with this drug in HMA failure. In this setting, a phase 1 study of atezolizumab plus azacitidine (#NCT02508870) and a phase 2 study of azacitidine plus ipilimumab or azacitidine plus nivolumab are ongoing (#NCT02530463). In MDS with HMA failure, ipilimumab as a single drug yielded an ORR of 30%, whereas nivolumab seemed ineffective.75 Another experimental study is combining decitabine with ipilimimab in HMA-refractory/relapsed MDS (#NCT02890329). More recently, combinations of various chemotherapy regimens (high- and low-dose regimens) with nivolumab are under evaluation (#NCT03259516) in MDS with HMA failure. A different clinical trial combining HDAC inhibitor entinostat with pembrolizumab is currently recruiting patients experiencing HMA failure irrespective of IPSS-R risk category (#NCT02936752).
The potential activity of anti-CD123 antibody talacotuzumab was evaluated in HMA-refractory/relapsed MDS patients, but the study was suspended (#NCT02992860) because of a lack of efficacy advantage and infusion-related adverse events.
New chemotherapy formulation
The liposomal formulation of cytarabine and daunomicine CPX-351, approved by the US Food and Drug Administration for advanced secondary AML and AML post-MDS, may improve tolerability and efficacy of classical high-dose chemotherapy, allowing treatment of MDS after HMA failure that has progressed to AML.79 A phase 2 study of CPX 351 in elderly patients with MDS and AML after HMA failure is currently recruiting (#NCT02019069).
Add-on strategy
In an attempt to restore responses to HMAs, maintenance of HMA treatment and addition of possible additive/synergistic agents were considered, under the hypothesis that the combination could rechallenge response, especially in secondary resistant patients. This was observed with lenalidomide added back into treatment in patients who had lost response to azacitidine.80 The same rationale is at the basis of recent studies combining HMAs with various agents possessing different mechanisms of action,81-83 but results have not been encouraging.84
Conclusions
Patients with HMA-refractory/relapsed MDS should always be considered for clinical experimental trials and encouraged to move to tertiary centers for cure. In fact, patients treated in experimental studies are followed closely, and aside from the activity of the tested drug, such close observation may itself improve outcome by improving supportive care. Consent is a particularly delicate issue for patients and caregivers; therefore, to increase compliance, sufficient time should be allocated to discuss details of the study, particularly randomization.
Many studies evaluating agents with various mechanisms of action have been and are being performed in MDS with HMA failure. Although some clearly have therapeutic potential, a majority of these agents are still in the early stages of development.
Without doubt, MDS patients for whom HMA therapy fails are a population with an extremely dismal outcome and with few therapeutic opportunities, whose difficult care burdens the health system.85 Proper management of first-line HMA therapy, with appropriate doses and prolonged treatment, may partially reduce primary resistance. Eventually, HMA-treated MDS patients lose response, and the best treatment to offer at present is inclusion in experimental trials. Selecting patients based on their cytogenetic and molecular characteristics at the moment of HMA failure may support the choice of personalized targeted therapy, already feasible for a certain number of typical alterations in MDS. Although sometimes difficult to organize for smaller centers and for elderly patients, enrollment in clinical studies would guarantee optimal care.
Acknowledgment
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC; IG 2015 Id 16861).
Authorship
Contribution: V.S. is responsible for all aspects of this article.
Conflict-of-interest disclosure: V.S. has received honoraria from Celgene, Janssen, and Novartis and served on advisory boards for Celgene, Janssen, AbbVie, Astex, Karyopharma, and Acceleron.
Correspondence: Valeria Santini, MDS Unit, AOU Careggi, University of Florence, Largo Brambilla 3, 50134 Firenze, Italy; e-mail: santini@unifi.it.
